
膵管腺がん(PDAC)を中心に、がん細胞集団の中でストレス顆粒(Stress Granules; SG)の量と質に大きな細胞間ヘテロ性が存在し、なかでもG2期細胞でSGが突出して増えること、そしてその駆動役がcPLA2 → 15d-PGJ2経路であることが明らかになりました。
本稿では、論文の主要結果を整理しつつ、開発の観点での示唆・実装の勘所・限界と次の一手を、SWELL投稿用の実務テンプレに沿って丁寧にまとめます。
目次
- 要点(まずはここだけ)
- 背景:SGと化学療法耐性
- 主な発見:G2で増えるSGと脂質メディエーター
- 作用機序:CASPASE-3 ⇄ cPLA2 ⇄ 15d-PGJ2 ⇄ SG
- エビデンスの層:in vitro / in vivo / 臨床材料
- 治療学的示唆:併用設計・患者層別化・PoC設計
- 計測・バイオマーカー:何を、いつ、どう測るか
- 創薬・開発の勘所:化合物設計と安全性の論点
- 限界と反証可能性:どこまで一般化できるか
- 実装ロードマップ:前臨床→臨床の具体ステップ
- FAQ:実務者からよく出る質問
1. 要点(まずはここだけ)
- SGは細胞周期依存で偏在し、G2期で最も多い。FUCCIを用いた単一細胞解析で、G2細胞の約9割がSG陽性、G1の数倍のSG指数。
- 脂質メディエーターの収束点は15d-PGJ2。G2で15d-PGJ2が10–20倍に上昇。外因性15d-PGJ2で非同期集団のSGがG2水準まで上昇。
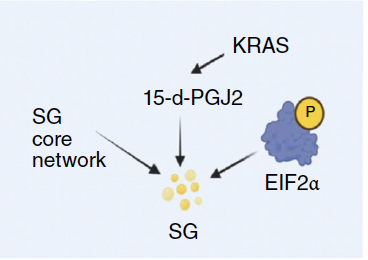
- 上流のスイッチはcPLA2。cPLA2活性がG2で顕著に高く、cPLA2阻害/ノックダウンでG2特異的SGが減少。15d-PGJ2添加でレスキュー。
- ブレーキ役はTR-cPLA2(cPLA2の切断断片)。G2ではCASPASE-3活性が低く、TR-cPLA2が減少→フル長cPLA2活性が上がる→15d-PGJ2↑→SG↑。
- 治療学的に重要:オキサリプラチンなどG2アレストを誘導する薬剤に対する適応的耐性としてSGが機能。cPLA2阻害剤との併用でSG低下・腫瘍増殖抑制を示す。
2. 背景:SGと化学療法耐性
SGは、翻訳抑制下でmRNAとRBPs(G3BP1/2、TIA-1、eIF群など)が液液相分離して形成される非膜オルガネラです。がん細胞では、SGがグローバルな翻訳負荷を軽減し、mRNAの一時退避やストレス応答経路の再配線を通じて薬剤耐性に寄与すると考えられています。ただし、従来研究は集団平均が中心で、細胞周期や微小環境に応じた細胞間ヘテロ性は十分に描けていませんでした。
PDACのような堅牢な腫瘍型では、一過性のG2アレストや酸化ストレス、低酸素、ERストレスなど実臨床に近い多様なストレスが重層化します。この状況下で、どの細胞が、いつ、どの程度SGを作るのかは、薬効と再増殖の分岐点になり得ます。
3. 主な発見:G2で増えるSGと脂質メディエーター
3.1 G2期でのSG偏在は普遍的な現象
- FUCCIで周期を可視化すると、G2がSG高負荷(G1の~6倍)。S期は中間。
- 過酸化水素、低酸素、ERストレスなどストレスの種類を問わず再現。
3.2 収束点は15d-PGJ2(PGD2由来)
- G2で15d-PGJ2が10–20倍に上昇。
- 外因性15d-PGJ2で、非同期集団のSG負荷をG2並みに増強。
- L-PGDS阻害(例:AT-56)で、G2/非同期の差が緩和し、PGD2→15d-PGJ2経路がSG形成の実効的な収束点であることを示唆。
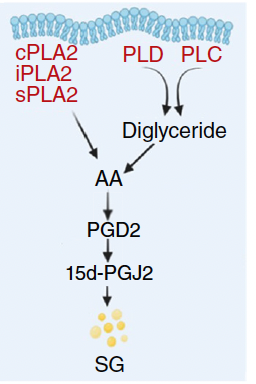
3.3 上流スイッチはcPLA2
- 脂質シグナルの上流群のうち、cPLA2活性のみがG2で顕著に上昇。
- cPLA2阻害剤(例:pyrrophenone)やcPLA2ノックダウンでG2特異的にSG減少。外因性15d-PGJ2でレスキュー。
4. 作用機序:CASPASE-3 ⇄ cPLA2 ⇄ 15d-PGJ2 ⇄ SG
4.1 TR-cPLA2は「内因性ブレーキ」
cPLA2には、CASPASE-3により切断される触媒能のない断片TR-cPLA2が存在し、これはフル長cPLA2の競合阻害因子として働きます。通常、適度なCASPASE-3活性によりTR-cPLA2が供給され、cPLA2活性にブレーキが掛かります。
4.2 G2ではCASPASE-3活性が低くブレーキが外れる
- G2期ではCASPASE-3活性が低下→TR-cPLA2が減少→フル長cPLA2活性が上昇。
- 結果としてPGD2 → 15d-PGJ2が増え、SG形成が促進。
4.3 因果性の補強
- CASPASE-3阻害で、非同期集団でもTR-cPLA2↓ / 15d-PGJ2↑ / SG↑が再現。
- 切断耐性cPLA2変異の発現でもcPLA2活性↑→SG↑が観察。
5. エビデンスの層:in vitro / in vivo / 臨床材料
5.1 in vitro(細胞レベル)
- FUCCI+SGマーカー(G3BP1など)で周期別SG負荷を定量。
- 脂質メディエーター測定で15d-PGJ2のG2選択的増加。
- 遺伝学的/薬理学的介入(cPLA2 KD、pyrrophenone、AT-56)で因果性を検証。
5.2 in vivo(動物モデル)
- PDAC正所性モデルや患者腫瘍移植で、Geminin陽性(S/G2)区画のSG負荷が高い。
- オキサリプラチン投与で一過性のG2偏りとSG上昇。
- cPLA2阻害+オキサリプラチン併用で、SG負荷低下と腫瘍増殖抑制が示唆。
5.3 臨床材料(探索)
- 患者腫瘍切片で、G3BP1×Gemininの二重染色がSG高負荷のS/G2分画を可視化。
- TR-cPLA2/cleaved-CASPASE-3比や15d-PGJ2は薬理動態・薬力学のバイオマーカー候補。
6. 治療学的示唆:併用設計・患者層別化・PoC設計
6.1 併用設計の原則
- G2アレスト誘導薬+cPLA2阻害剤の時限式併用で、SG依存の適応耐性を断ち切る。
- cPLA2単剤はG2バイアスが低い状態では効きにくいため、周術期や維持療法というより細胞周期偏りが出る局面に同期。
- 15d-PGJ2測定やSG/Geminin計測でタイミング調整。
6.2 患者層別化(誰に効く?)
- ベースラインでSG負荷が高い腫瘍(G3BP1高発現、Geminin高分画)。
- 治療導入後にG2偏りや15d-PGJ2上昇が検出される症例。
- 翻訳ストレス経路(PERK/eIF2α/ISR)活性が背景にある腫瘍。
6.3 PoCの指標
- 組織/リキッドでの15d-PGJ2、脂質オミクス。
- G3BP1×Geminin IHCによるSG高負荷分画の比率変化。
- TR-cPLA2/cleaved-CASPASE-3比の推移。
- 腫瘍縮小ではなくとも増殖速度の鈍化・再増殖遅延を見るエンドポイント設計。
7. 計測・バイオマーカー:何を、いつ、どう測るか
7.1 何を測るか
- SGそのもの:G3BP1、TIA-1などの粒子数/面積(IF)
- 細胞周期:Geminin(S/G2)、Ki-67、pH3(M)
- 脂質:15d-PGJ2(LC-MS/MS)、PGD2、AA代謝物
- スイッチ:cPLA2活性、TR-cPLA2量、CASPASE-3活性
7.2 いつ測るか
- 導入前:ベースラインのSG/Geminin層別化
- 初回化療後の早期:G2偏りピークを狙い脂質・SGを再計測
- 併用開始後:SG負荷低下と脂質メディエーターのダイナミクスを追跡
7.3 どう測るか(実務TIPS)
- 組織IFはコアラボと標準化。粒子解析(数・サイズ・クラスタリング)は前処理が結果を大きく左右。
- 脂質測定は安定同位体内部標準を厳密運用。サンプル調製と保管温度を徹底。採血から安定化までの時間もSOP化。
8. 創薬・開発の勘所:化合物設計と安全性の論点
8.1 cPLA2阻害剤の要件
- 選択性:他PLA2(sPLA2, iPLA2)やCOX/LOXへのオフターゲット回避。
- ADMET:腫瘍移行性、肝代謝、薬物相互作用(特に化療との併用時)。
- 剤形:急性併用に耐えるPK(Cmax/Tmax)と安全域。
8.2 安全性プロファイル
- cPLA2は炎症・免疫・神経にも関与。全身阻害は感染リスクや創傷治癒への影響を評価。
- 15d-PGJ2は求核性の高いα,β不飽和ケトン。プロテオームへの共有結合修飾が広がる可能性→系統毒性に留意。
8.3 併用設計の薬理
- 時間合わせ(タイムド・コンビネーション):化療投与→G2バイアス出現→短時間でcPLA2阻害を重ねる。
- 量合わせ(エクスポージャー最適化):化療の用量制限毒性(DLT)を超えない範囲でcPLA2阻害の曝露を最大化。
9. 限界と反証可能性:どこまで一般化できるか
- 腫瘍型の多様性:PDAC以外での再現性(大腸・胃・肺・乳など)を系統比較。
- G2非依存の耐性機構:例えばDNA修復経路(HR/NHEJ)や薬剤排出の寄与を分離検証。
- 微小環境:低酸素・酸化ストレス・栄養ストレスがSGをどこまで増幅するか。
- 適応耐性の置き換え:SGを抑えると、別経路(オートファジー、UPR、ISRIB感受性など)が立ち上がる可能性。
10. 実装ロードマップ:前臨床→臨床の具体ステップ
10.1 前臨床(6–12か月)
- モデル確立:PDAC PDO/PDXでG2バイアス→SG増→併用でのSG低下・腫瘍抑制を再現。
- PK/PD連結:cPLA2阻害剤の血中・腫瘍内曝露と15d-PGJ2・SGの変動を連結。
- 安全性:短期併用毒性(骨髄・肝・腎・消化管・感染感受性)。
10.2 早期臨床(PoC)
- 対象:再発/切除不能PDAC、オキサリプラチン併用レジメン下で層別化。
- デザイン:ランダム化Phase Ib/II。バイオマーカー層別+適応的デザイン。
- 主要評価:SG負荷変化(IF)、15d-PGJ2、無増悪期間(PFS)、早期の再増殖遅延。
11. FAQ:実務者からよく出る質問
Q1. cPLA2単剤は効かないの?
G2偏りが小さい局面では効果が限定的。G2を作る化療や放射線と組み合わせ、時間同期させるのがポイントです。
Q2. 15d-PGJ2を外から足せばよい?
15d-PGJ2は広範なタンパク修飾を起こし得るため治療薬としては不向き。上流のcPLA2/PGD2経路を制御するほうが現実的です。
Q3. SGを直接壊す薬は?
SG形成の直接阻害は翻訳・ストレス応答全体を揺らすリスクが高い。脂質メディエーター側からの調整は、G2細胞に偏在する“脆弱なスイッチ”を狙える点が長所です。
12. 私の考察:創薬につながるストーリー
本研究の核は、「SGは均一にできるわけではない」という事実を、細胞周期という分かりやすい軸で説明し、脂質メディエーターという介入可能なノードに落とし込んだ点です。臨床的に重要なのは、SGが“常に悪”ではなく“状況依存の適応盾”であること。したがって、盾を構えるタイミング(G2偏り)を検出し、その瞬間だけcPLA2で盾を外す——このタイムド・コンビネーションこそが戦略の肝になります。
今後の拡張としては、腫瘍型横断の層別化クライテリア(SG/Geminin指標+脂質プロファイル)を規格化し、ISR(統合ストレス応答)やオートファジーなど隣接経路との二段・三段の多層併用を設計すること。さらに、腫瘍微小環境(低酸素・酸化ストレス)が脂質経路に与える影響を定量化すれば、放射線療法のようなG2バイアスを作る治療との相性評価も前に進みます。
要するに、「G2でSGが立つ」→「cPLA2で倒す」という状況依存の脆弱性は、PDACのような難治がんにおける再増殖の“わずかな綻び”を突く有望な手段に見えます。臨床級のcPLA2阻害剤、正しく設計されたPDバイオマーカー、時間同期設計——この三点が揃えば、耐性破りの現実解に手が届くと考えます。
13. 参考文献
- Redding A., et al. Cancer Discovery, 2025. Cytosolic Phospholipase A2 Determines Intercellular Heterogeneity of Stress Granules and Chemotherapy Response.(詳細は原著論文参照)






この記事はMorningglorysciencesチームによって編集されました。
※本記事は学術研究の要点を実務者目線で再構成したものであり、診療ガイドではありません。治療方針は必ず担当医とご相談ください。






コメント