
Introduction: A New Relationship Between Transcription Factors and DNA Repair
Recent cancer research has highlighted DNA replication errors and defects in repair mechanisms as central drivers of tumorigenesis. The mismatch repair (MMR) system, in particular, is known to be a key safeguard against cancer by correcting replication errors. On the other hand, transcription factors (TFs) are essential regulators of gene expression, but their potential to interfere with DNA repair has been underappreciated.
What you will learn
- Introduction: A New Relationship Between Transcription Factors and DNA Repair
- DNA Replication Errors and Repair Basics
- Previous Evidence of TF-Repair Interference
- New Findings from the Cell Study
- High Transcriptional Activity and Tumor Progression
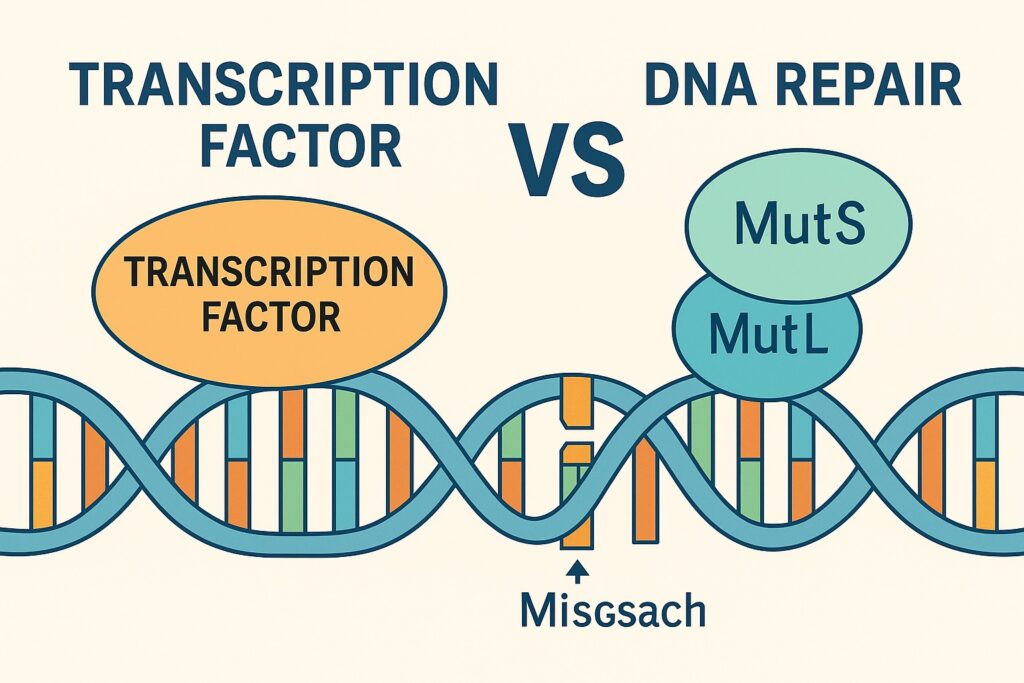
A study published in Cell in July 2025, titled “DNA mutagenesis driven by transcription factor competition with mismatch repair,” revealed a novel mechanism: transcription factors compete with the MMR factor MutSα, thereby preventing recognition of DNA mismatches and driving high-frequency mutations at TF binding sites. This finding suggests that transcriptional activity not only regulates gene expression but may also induce mutations that fuel tumor progression and malignancy.
DNA Replication Errors and Repair Basics
High Fidelity and Its Limits
DNA replication in eukaryotic cells is highly faithful, with only a handful of errors per several billion nucleotides per division. Yet, replication mistakes do occur. If unrepaired, they become fixed mutations that may drive cancer and genetic diseases.
The Role of Mismatch Repair (MMR)
The mismatch repair system identifies and corrects errors such as base mispairs and small insertions/deletions after DNA replication. MutSα (the MSH2-MSH6 complex) plays a central role by recognizing mismatches and initiating repair. When this system fails, microsatellite instability (MSI) arises, leading to dramatically increased cancer risk.
Previous Evidence of TF-Repair Interference
Earlier studies showed that transcription factor binding can block nucleotide excision repair of UV-induced lesions (Sabarinathan et al., Nature 2016). Similarly, binding sites for CTCF and cohesin were found to be hotspots for mutations in cancer genomes (Katainen et al., Nat Genet 2015). These observations suggested that TFs may act as physical obstacles to DNA repair machinery.
New Findings from the Cell Study
Study Overview
The recent Cell study used yeast genetic assays to show that TFs directly compete with MutSα, preventing mismatch recognition and causing mutations to accumulate at TF binding sites. This phenomenon was observed across multiple TF families and MutSα homologs, indicating a conserved mechanism.
Analysis in Human Cancer
Analysis of human cancer genomes confirmed elevated mutation rates at TF binding sites, particularly those bound by the oncogenic transcription factor MYC. Notably, these mutations were enriched even in MMR-proficient cancers, indicating that TF binding itself can hinder repair activity.
High Transcriptional Activity and Tumor Progression
MYC is a canonical oncogene that amplifies transcription globally, driving cell growth and metabolism. The study showed that high transcriptional activity can clash with repair processes, leading to mutation accumulation. Thus, MYC activation is not only a fuel for tumor growth but also a generator of mutations that may accelerate malignancy.
Neoantigen Formation and Immune Response
Some of these mutations create novel peptide sequences, or neoantigens, that can be recognized by the immune system as foreign. Incomplete MMR function accelerates mutation accumulation, producing abundant neoantigens. This principle underlies the remarkable responsiveness of MSI-high (MSI-H) tumors to immune checkpoint inhibitors (ICIs).
Clinical Case: MSI-H Colorectal Cancer and Lynch Syndrome
A pivotal clinical trial conducted at Memorial Sloan Kettering Cancer Center (Le et al., NEJM 2017) demonstrated striking efficacy of PD-1 blockade in patients with mismatch repair-deficient (MSI-H) colorectal cancer, including those with Lynch syndrome. The trial showed that MMR deficiency–driven mutational burden and neoantigen formation directly translate into powerful therapeutic vulnerability.
The new findings in Cell resonate strongly with this success: TF-induced impairment of MMR may also fuel neoantigen generation, thereby enhancing immune responses against tumors.
Discussion: Toward a New Therapeutic Model
This study highlights the dual role of TF binding–induced mutations in cancer. On the one hand, they promote tumor progression and malignancy; on the other, they enrich the pool of neoantigens, enhancing responsiveness to immunotherapy. The challenge now is to determine which tumor types may benefit most from exploiting this mechanism clinically.
Colorectal cancer with MSI-H and Lynch syndrome is a clear example, but tumors with high MYC activity or other transcriptional amplifiers may also prove susceptible to immunotherapy through this pathway.
Conclusion and Outlook
The competition between transcription factors and DNA repair represents a previously overlooked driver of mutagenesis. This mechanism deepens our understanding of tumorigenesis while opening new opportunities for immunotherapy based on neoantigen exploitation. Future cancer research and therapy development will increasingly need to focus on the balance between transcriptional activity and DNA repair fidelity.
References
- Wei Zhu et al. Cell, 2025. “DNA mutagenesis driven by transcription factor competition with mismatch repair”
- Sabarinathan R. et al. Nature, 2016. “Nucleotide excision repair is impaired by binding of transcription factors to DNA”
- Katainen R. et al. Nature Genetics, 2015. “CTCF/cohesin-binding sites are frequently mutated in cancer”
- Le DT. et al. New England Journal of Medicine, 2017. “Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade”
- Gabay M. et al. Cold Spring Harb Perspect Med, 2014. “MYC activation is a hallmark of cancer initiation and maintenance”
This article was edited by the Morningglorysciences team.

Comments