
Human Brain Organoids Reveal Therapeutic Potential for Rare Pediatric Epilepsy
In June 2025, a research team led by Dr. Heather Mefford at St. Jude Children’s Research Hospital reported a groundbreaking study using human brain organoids to model and treat a rare pediatric disorder known as UBA5-associated epileptic encephalopathy. Published in Science Translational Medicine, the study sheds light on how patient-derived organoids can recapitulate disease features and guide therapeutic strategies for intractable childhood neurological conditions. This work stands out for using only human-derived cells—without animal models—to bridge fundamental biology and translational therapy.
UBA5 Mutations Disrupt Protein Modification and Neuronal Stability
UBA5 encodes an enzyme essential for ufmylation, a ubiquitin-like post-translational modification system that regulates cellular homeostasis. Patients in this study carry compound heterozygous mutations—one loss-of-function allele and one partially functional allele—resulting in significantly reduced UBA5 activity. This reduction impairs ufmylation-dependent processes critical for neurodevelopment. In particular, defective ufmylation compromises inhibitory signaling in the developing brain, leading to hyperexcitability and severe epilepsy. This mechanistic insight into how subtle changes in protein modification cascade into major network disruptions marks a leap in understanding pediatric epileptogenesis.
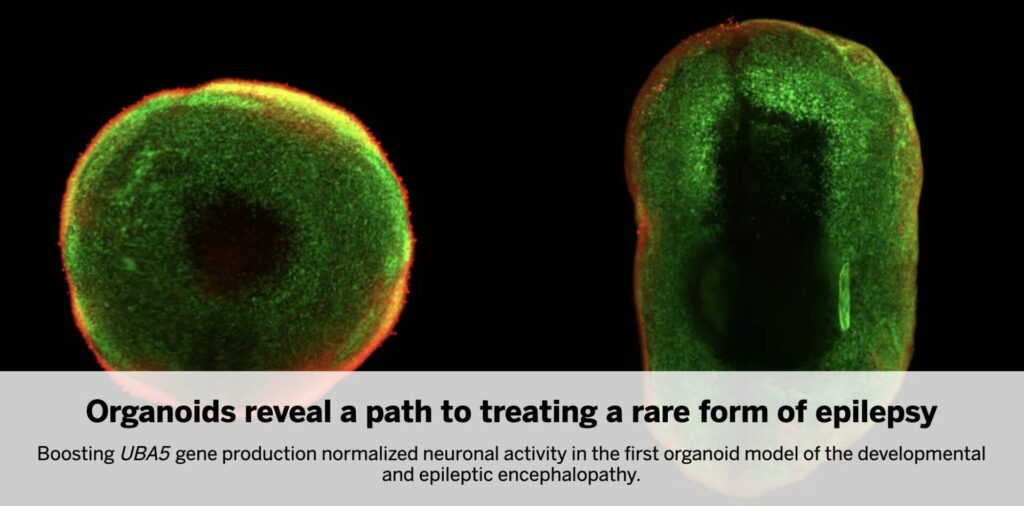
Patient-Derived Organoids Model Structural and Functional Deficits
To model disease pathogenesis, the team generated cerebral organoids from induced pluripotent stem cells (iPSCs) derived from the patient and their parents. The patient’s organoids were approximately 25% smaller, with delayed cortical-like layering and reduced complexity. Most notably, they exhibited a marked reduction in GABAergic inhibitory neurons—a population critical for balancing neural excitation. Electrophysiological recordings revealed abnormal firing patterns and spontaneous network discharges, mirroring the clinical phenotype of the patient. This functional recapitulation validates the organoid as a faithful model and highlights its translational value for rare neurodevelopmental diseases.
Therapeutic Rescue via UBA5 Upregulation Strategies
The researchers tested two approaches to increase UBA5 expression in organoids: (1) CRISPR activation (CRISPRa) targeting the endogenous UBA5 promoter, and (2) a long non-coding RNA-based enhancer called SINEUP, which improves mRNA translation. Both interventions successfully increased UBA5 protein levels, restored ufmylation signaling, and normalized neuronal activity. While SINEUP effects were transient, the study provides strong proof-of-concept that even modest increases in residual UBA5 activity can reverse cellular phenotypes. This lays the groundwork for developing targeted therapeutics—either gene therapy or small molecule-based—that enhance partial-function alleles.
Broader Implications and Future Directions
This work opens several avenues for translational and mechanistic research:
- Investigating why inhibitory neurons are disproportionately vulnerable to UBA5 dysfunction
- Evaluating long-term safety and efficacy of CRISPRa or SINEUP in vivo
- Exploring therapeutic windows during early brain development
- Optimizing delivery systems for sustained expression (e.g., viral vectors, RNA nanoparticles)
- Assessing generalizability of the organoid approach to other rare epileptic encephalopathies
My Insight
This study exemplifies the power of human-only organoid systems to model and treat complex pediatric diseases. Without relying on animal models, the researchers uncovered a mechanistic framework and therapeutic concept for a devastating condition. For the future of rare disease drug development, organoids are not just a model—they are a platform for clinical innovation.

Comments