
Across pancreatic ductal adenocarcinoma (PDAC) models, tumor cells show marked intercellular heterogeneity in stress granules (SGs). A single-cell, cell-cycle–aware view reveals that SGs are maximally enriched in G2, powered by a lipid mediator axis cPLA2 → PGD2 → 15d-PGJ2. In G2, lower non-apoptotic CASPASE-3 activity reduces an endogenous inhibitory fragment of cPLA2 (TR-cPLA2), thereby disinhibiting cPLA2, boosting 15d-PGJ2, and amplifying SG formation. Therapeutically, cPLA2 blockade suppresses G2-skewed SGs and enhances the efficacy of G2-arresting chemotherapy (e.g., oxaliplatin). This article distills the study’s logic and translates it into development-ready insights for biomarker strategy, combination design, and PoC planning.
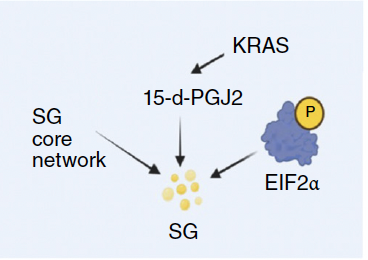
Table of Contents
- Key Points (TL;DR)
- Background: SGs and Therapy Resistance
- Core Findings: G2-Enriched SGs and Lipid Mediators
- Mechanism: CASPASE-3 ⇄ cPLA2 ⇄ 15d-PGJ2 ⇄ SG
- Evidence Layers: in vitro / in vivo / Clinical Specimens
- Therapeutic Implications: Combinations & Patient Stratification
- Measurement & Biomarkers: What / When / How
- Development Notes: Chemistry & Safety
- Limitations & Falsifiability
- Implementation Roadmap: Preclinical → Clinical
- FAQ for Practitioners
1. Key Points (TL;DR)
- SGs are cell-cycle dependent and peak in G2. FUCCI-based single-cell analyses show ~90% of G2 cells SG-positive with an SG index several-fold higher than G1.
- 15d-PGJ2 is the convergent lipid mediator. G2 cells display a 10–20× surge of 15d-PGJ2. Exogenous 15d-PGJ2 elevates SG load in asynchronous populations to G2-like levels.
- cPLA2 is the upstream switch. G2-selective elevation of cPLA2 activity is necessary for SG amplification; cPLA2 inhibition/knockdown reduces SGs in G2, rescued by 15d-PGJ2.
- TR-cPLA2 acts as an endogenous brake. G2 shows lower CASPASE-3 activity, reducing TR-cPLA2 and thus disinhibiting full-length cPLA2 → 15d-PGJ2↑ → SG↑.
- Clinical angle: SGs encode an adaptive resistance to G2-arresting drugs (e.g., oxaliplatin). cPLA2 inhibitors lower SG burden and sensitize tumors under G2 bias.
2. Background: SGs and Therapy Resistance
Stress granules emerge under translational repression when mRNAs and RBPs (e.g., G3BP1/2, TIA-1, eIFs) undergo liquid–liquid phase separation. In cancer, SGs are viewed as adaptive hubs buffering proteostasis and stress signaling, thereby shaping drug tolerance and regrowth. Yet most datasets average across populations, masking cell-to-cell heterogeneity linked to microenvironmental cues and the cell cycle. PDAC—being hypoxic, nutrient-stressed, and therapy-exposed—offers a natural testbed to interrogate who builds SGs, when, and how much.
3. Core Findings: G2-Enriched SGs and Lipid Mediators
3.1 G2 enrichment is robust across stressors
- FUCCI demarcation shows a G2-high SG phenotype (≈6× vs. G1); S-phase sits in between.
- The pattern holds across oxidative, ER, and hypoxic stress, implying a generalizable logic.
3.2 15d-PGJ2 is the operative convergence point
- G2 cells exhibit order-of-magnitude elevation of 15d-PGJ2.
- Exogenous 15d-PGJ2 recapitulates high-SG states in asynchronous cultures.
- Inhibition of L-PGDS (e.g., AT-56) blunts the differential, indicating a PGD2 → 15d-PGJ2 axis as a functional bottleneck for SG formation.
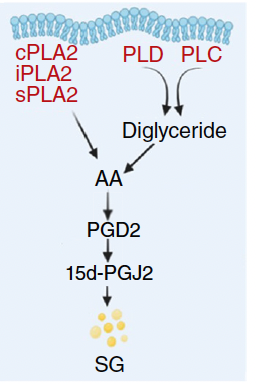
3.3 cPLA2 is the G2-specific upstream driver
- Among lipid-signaling candidates, only cPLA2 activity rises prominently in G2.
- cPLA2 inhibition (e.g., pyrrophenone) or cPLA2 knockdown markedly reduces SG burden in G2; 15d-PGJ2 fully rescues.
4. Mechanism: CASPASE-3 ⇄ cPLA2 ⇄ 15d-PGJ2 ⇄ SG
4.1 TR-cPLA2 is an intrinsic brake
cPLA2 can be cleaved by CASPASE-3 to yield a catalytically inactive fragment (TR-cPLA2) that competitively inhibits full-length cPLA2. Under “baseline” non-apoptotic CASPASE-3 activity, this fragment restrains cPLA2 and keeps lipid signaling in check.
4.2 G2 lifts the brake by lowering CASPASE-3 activity
- G2 shows reduced non-apoptotic CASPASE-3 → TR-cPLA2 decreases → full-length cPLA2 disinhibited.
- Consequently, PGD2 → 15d-PGJ2 rises and SG assembly is promoted.
4.3 Causality stress-tested
- Pharmacologic CASPASE-3 inhibition in asynchronous cells recapitulates TR-cPLA2↓ / 15d-PGJ2↑ / SG↑.
- Expression of a cleavage-resistant cPLA2 variant similarly boosts cPLA2 activity and SGs.
5. Evidence Layers: in vitro / in vivo / Clinical Specimens
5.1 in vitro
- FUCCI + SG markers (e.g., G3BP1) enable cycle-resolved SG quantification.
- Lipidomics shows G2-selective increases in 15d-PGJ2.
- Genetic/pharmacologic interrogation (cPLA2 KD, pyrrophenone, AT-56) establishes necessity/sufficiency.
5.2 in vivo
- Orthotopic PDAC and patient-derived tumors show a Geminin-positive (S/G2) compartment with high SG load.
- Oxaliplatin transiently shifts the cycle toward G2 and increases SGs.
- cPLA2 inhibitor + oxaliplatin co-treatment reduces SGs and slows tumor growth compared with monotherapy.
5.3 Clinical materials (exploratory)
- Dual IHC (G3BP1 × Geminin) visualizes SG-high S/G2 fractions in human specimens.
- TR-cPLA2 / cleaved-CASPASE-3 ratios and 15d-PGJ2 levels emerge as PD biomarkers to track on-target engagement.
6. Therapeutic Implications: Combinations & Patient Stratification
6.1 Principles of combination design
- Use G2-arresting agents + cPLA2 inhibitors in a timed combination to cripple SG-mediated adaptive tolerance.
- Expect limited cPLA2 monotherapy benefit without a G2 bias; prioritize windows with cycle skew (e.g., post-chemo surge).
- Leverage 15d-PGJ2 kinetics and SG/Geminin readouts to schedule exposure.
6.2 Who is likely to benefit?
- Tumors with high baseline SG burden (G3BP1-high) and a sizable Geminin-positive fraction.
- Cases that increase G2 bias and 15d-PGJ2 upon therapy initiation.
- Background activation of translational stress pathways (e.g., PERK/eIF2α/ISR).
6.3 PoC endpoints to track
- 15d-PGJ2 in tissue or liquid biopsy; orthogonal lipid panels.
- G3BP1 × Geminin IHC—fractional changes in SG-high S/G2 compartments.
- TR-cPLA2 / cleaved-CASPASE-3 as a mechanistic PD anchor.
- Beyond shrinkage: growth-rate slowing, time-to-regrowth as sensitive readouts.
7. Measurement & Biomarkers: What / When / How
7.1 What to measure
- SGs: particle count/area of G3BP1, TIA-1 by IF.
- Cell cycle: Geminin (S/G2), Ki-67, phospho-H3 (M).
- Lipids: 15d-PGJ2, PGD2, AA metabolites (LC-MS/MS).
- Switch nodes: cPLA2 activity, TR-cPLA2 abundance, CASPASE-3 activity.
7.2 When to measure
- Baseline: SG/Geminin stratification to define starting state.
- Early on-treatment: capture the G2-skew peak to reassess lipids and SGs.
- After combo onset: verify SG decline and lipid mediator dynamics.
7.3 How to measure (practical notes)
- Standardize IF workflows with the core lab; particle analysis is sensitive to pre-processing.
- Stabilize lipidomics rigorously with isotope-labeled internal standards, temperature control, and time-to-quench SOPs.
8. Development Notes: Chemistry & Safety
8.1 What a clinical cPLA2 inhibitor must achieve
- Selectivity: avoid sPLA2/iPLA2 and COX/LOX off-targets.
- ADMET: tumor penetration, hepatic metabolism, DDI profile (especially with chemo).
- PK shape: exposure aligned to short windows of G2 bias (Cmax/Tmax fit for timed combos).
8.2 Safety landscape
- cPLA2 touches inflammation, immunity, and neural circuits; whole-body inhibition warrants infection/wound-healing vigilance.
- 15d-PGJ2 is an electrophile (α,β-unsaturated ketone) with broad protein adduction potential → monitor systemic toxicity.
8.3 PK/PD choreography for combinations
- Timing: chemo dosing → G2 swell → rapid cPLA2 blockade during this window.
- Exposure: maximize cPLA2 engagement without exceeding chemo DLTs; consider short, high-exposure pulses.
9. Limitations & Falsifiability
- Across tumor types: map reproducibility beyond PDAC (CRC, gastric, lung, breast) with standardized SG/cycle assays.
- Cycle-independent resistance: disentangle contributions from DNA repair (HR/NHEJ), drug efflux, or ISR rewiring.
- Microenvironment: quantify how hypoxia/oxidative/nutrient stress magnify SGs and lipid fluxes.
- Adaptive rerouting: expect compensations (e.g., autophagy, ISRIB sensitivity); design second-layer combos.
10. Implementation Roadmap: Preclinical → Clinical
10.1 Preclinical (≈6–12 months)
- Modeling: PDAC PDO/PDX to replicate the G2→SG surge and its cPLA2-inhibitor reversal.
- PK/PD linkage: correlate tumor exposure of cPLA2 inhibitor with 15d-PGJ2 and SG dynamics.
- Safety: short-course combo tox (marrow, liver, kidney, GI, infection risk).
10.2 Early clinical PoC
- Population: unresectable/recurrent PDAC under oxaliplatin-containing regimens with biomarker-based stratification.
- Design: randomized phase Ib/II with adaptive features; embed SG/Geminin and lipid PD.
- Endpoints: SG-burden shift (IF), 15d-PGJ2 kinetics, PFS, and time-to-regrowth.
11. FAQ for Practitioners
Q1. Is cPLA2 monotherapy useful?
Generally limited without a G2-skewed window. Pair with G2-arresting modalities and time the exposure.
Q2. Why not dose 15d-PGJ2 directly?
It is chemically reactive and promiscuous; controlling the upstream cPLA2/PGD2 axis is more tractable and specific.
Q3. Are there SG-directed drugs?
Direct SG disruption risks broadly perturbing translation and stress responses. The lipid-mediated, G2-biased switch offers a context-selective handle with fewer global liabilities.
12. Author’s Perspective: A Drug Discovery Narrative
The study’s central advance is to reframe SG biology through a cell-cycle lens and anchor it to a drug-gable lipid node. SGs are not uniformly “bad”; they are a situational shield. The actionable takeaway is temporal precision: detect when the shield is raised (G2 bias), and then take it down (cPLA2 inhibition). This is the essence of a timed combination.
To scale beyond PDAC, we should formalize cross-tumor criteria—SG/Geminin IHC plus lipidomics—and engineer second-layer combinations (ISR/autophagy modulators) for adaptive rerouting. Quantifying how hypoxia and redox stress feed the lipid axis will also clarify synergies with radiation or other cycle-skewing therapies. In short: “SGs rise in G2” → “disarm via cPLA2” reveals a conditional vulnerability that can be exploited with the right biomarkers, the right inhibitor, and the right timing.
13. References
- Redding A., et al. Cancer Discovery, 2025. Cytosolic Phospholipase A2 Determines Intercellular Heterogeneity of Stress Granules and Chemotherapy Response. (See the original article for figures and full methods.)
PDAC related articles






This article was edited by the Morningglorysciences team.






Comments