Key Takeaways
- Building on Volume 1’s preview, this article unpacks the most important convergent finding across the three April 2026 Nature Medicine trials: FMT works because patients lose their own deleterious bacteria — not because they acquire beneficial bacteria from the donor. We dive deep into the FMT-LUMINate analysis (Elkrief et al., Nat Med 2026;32:1337-1350).
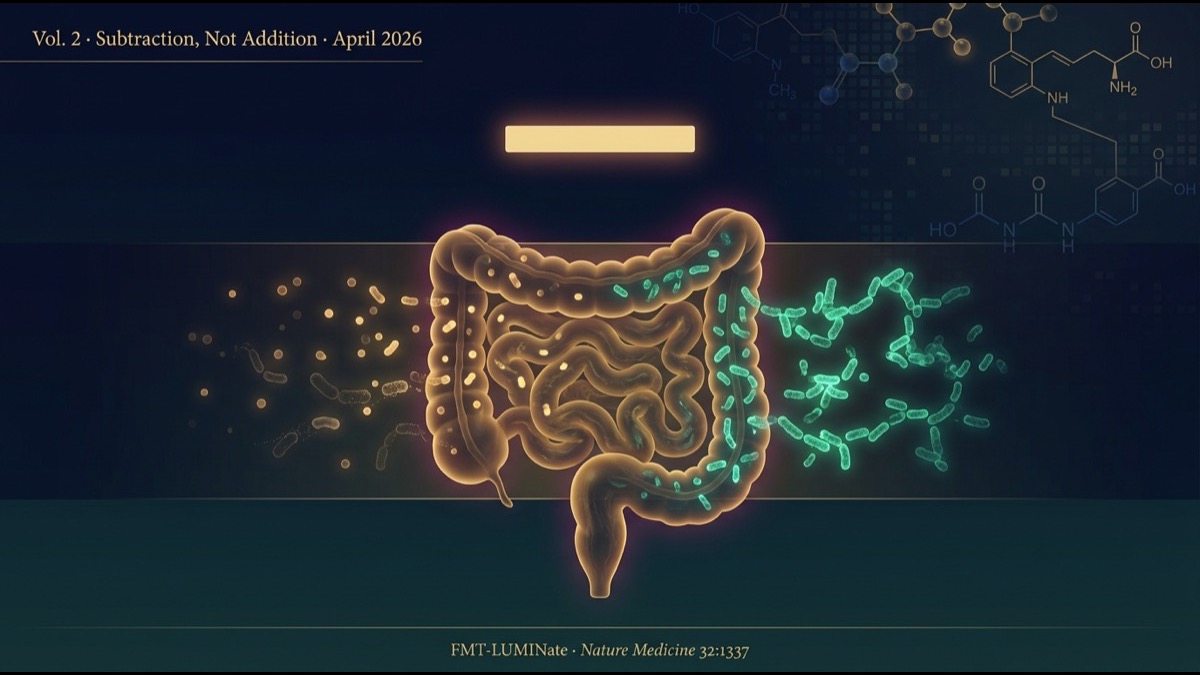
- Responders and non-responders showed no significant difference in donor-strain engraftment — α-diversity, β-diversity, Bray−Curtis dissimilarity, and StrainPhlAn all aligned. What separated them was whether known “bad-actor” taxa — Enterocloster citroniae, Clostridium innocuum, Enterocloster lavalensis — selectively disappeared.
- The team performed a reverse experiment: in antibiotic-pretreated mice given a responder’s stool, reintroducing the species that had been lost abolished the antitumor effect (P=0.028, P=0.022). Causality was proven.
- Mechanistically, deleterious bacteria sustain a tryptophan-pathway disturbance (kynurenine/quinolinic acid) that maintains an immunosuppressive milieu. Their loss releases CD8⁺ T-cell activation and reduces regulatory T-cells, freeing ICI activity.
- Implications are decisive: FMT/LBP design is shifting from addition to subtraction. Phage therapy, narrow-spectrum antibiotics, and competitive-exclusion consortium LBPs are entering meaningful clinical phases in 2026–27.
Introduction — A 10-Year Hypothesis Overturned by One Paper
For a decade — since Vétizou et al. ( Science ) in 2015 — the scientific community’s discussion of cancer immunotherapy and the gut microbiome has rested on an implicit premise: “Deliver good bacteria to the host, and the ICI will work better.”
That premise was powerful. In mice, transplanting human stool into germ-free or antibiotic-treated animals could overcome ICI resistance. Bifidobacterium, Akkermansia muciniphila, Faecalibacterium prausnitzii — papers spotlighting specific “good” species filled Science, Nature, and Cell Host & Microbe.
Drug companies followed suit. Seres Therapeutics, Vedanta Biosciences, Exeliom Biosciences and other US/EU ventures pursued “good-bacteria consortia” as live biotherapeutic products (LBPs). “Which good bacteria to pick, how to culture them, how to deliver them” was the central battlefield of the past decade.
April 2026 shook the premise. The core paper FMT-LUMINate (Elkrief et al., Nat Med 2026;32:1337-1350) showed that what separated ICI responders from non-responders was not “what was acquired from the donor” but “what was lost from oneself.” And the causal claim was directly proven in mouse experiments.
This article walks through that “subtraction” finding step by step for first-time readers, and then digs into the biochemistry and immunology of the mechanism. We close with how this discovery is reshaping next-generation FMT/LBP development.
Main Body
1. “Move Good Bacteria from the Donor” — The 10-Year Hypothesis
Before the main argument, a quick refresher on FMT’s logic.
Fecal microbiota transplantation (FMT) transfers stool from a healthy donor into the recipient’s gut, “resetting” or “overwriting” the microbiome. For recurrent Clostridioides difficile infection, where antibiotics cannot eradicate the pathogen, FMT — the idea of pushing out the offender with a healthy community — is now standard care.
For cancer immunotherapy, the expected logic was: “Patients with poor or imbalanced microbiomes lack the immune activation that ICIs require. Transplanting a healthy donor’s rich microbiome supplies the missing bacteria, immunity wakes up.” A textbook “addition” model.
On top of that model, the past decade accumulated experimental support:
- In mice, “adding” Bacteroides fragilis or Akkermansia muciniphila rescued or boosted anti-CTLA-4 / anti-PD-1 efficacy.
- In human cohorts, responders’ stool was enriched for response-associated taxa (Ruminococcaceae, Faecalibacterium, etc.).
- Patients given antibiotics shortly before ICI showed lower response rates — disrupt the microbiome, lose efficacy.
All evidence pointed in the same direction: “good bacteria present → drug works; missing → it doesn’t.” FMT trials were designed to test this.
2. What FMT-LUMINate Really Was — What “Non-Responders'” Data Said
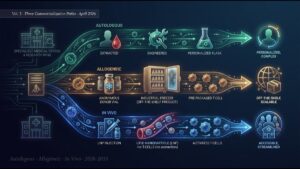
FMT-LUMINate enrolled 20 treatment-naive NSCLC patients (PD-L1 ≥ 50%) and 20 treatment-naive cutaneous melanoma patients, each receiving a single oral-capsule FMT before ICI initiation. As detailed in Volume 1, the headlines were striking: NSCLC ORR 80%, melanoma ORR 75%, both well above historical benchmarks and meeting the trial’s primary endpoint.
Volume 2 dives into the deeper question: what differed in the gut microbiomes of responders (R) versus non-responders (NR)? The team layered shotgun metagenomics, qPCR, high-throughput culturomics, plasma metabolomics, and peripheral-blood flow cytometry to attack this question from multiple independent angles.
What emerged is the opposite of the “addition” hypothesis.
3. The Diversity Trap — α/β Diversity Doesn’t Predict Response
The first stop in microbiome analysis is diversity metrics.
α-diversity (alpha) measures the number and evenness of species within a sample (Shannon index, etc.). The folk wisdom: a healthier gut has higher α-diversity.
β-diversity (beta) measures compositional differences between samples (Bray−Curtis dissimilarity, etc.). For FMT, it captures “how donor-like did the recipient become?” or “how much did the patient drift from baseline?”
Comparing FMT-LUMINate’s R and NR groups produced a striking result.
| Metric | NSCLC | Melanoma | Interpretation |
|---|---|---|---|
| Baseline α-diversity (R vs NR) | No difference | No difference | “Microbiome richness” doesn’t predict response |
| Post-FMT α-diversity (R vs NR) | No difference | No difference | Same after FMT |
| Baseline β-diversity (R vs NR) | P=0.4 | P=0.9 | Original composition is similar between R and NR |
| Post-FMT β-diversity (R vs NR) | P=0.09 | P=0.006 | Some shift after FMT — but what is it? |
Diversity numbers and overall composition cannot explain why the drug works for some and not for others. The signal had to live at finer resolution.
4. Donor-Strain Engraftment Was Identical Between Responders and Non-Responders
The team escalated the resolution.
Using the StrainPhlAn pipeline, they tracked engraftment at the strain (subspecies) level rather than the species level. Specifically, they compared Bray−Curtis dissimilarity (= how close did the recipient drift toward the donor?) and the absolute number of donor-derived strains, between R and NR.
The result was a direct refutation of the prior model.
- Bray−Curtis dissimilarity: no significant difference between R and NR. “Did you become like the donor?” did not determine response.
- Donor-derived strain count: no significant difference. “How much donor bacteria engrafted?” also did not determine response.
This is a sharp blow to the decade-long “deliver good bacteria → response” model. At least in FMT-LUMINate, something other than donor-derived bacteria was driving the outcome.
5. Finding “Subtraction” — Selective Loss of Deleterious Bacteria
So what is that “something else”? The team tracked species-level genome bins (SGBs) over time, classified into five categories:
- Bacteria from the donor that were absent at baseline in the patient (= textbook engraftment)
- Bacteria unique to the patient at baseline
- Bacteria common to both patient baseline and donor
- Bacteria present at patient baseline but not detected after FMT (= lost bacteria)
- New bacteria absent from both patient baseline and donor that appeared after FMT
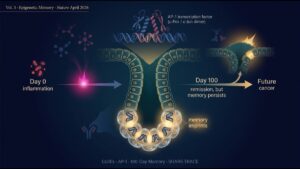
Comparing R vs NR across these five categories, the decisive difference appeared in category 4 (lost bacteria).
Responders lost significantly more of their own baseline taxa than non-responders (P=0.016). NSCLC subgroup alone was significant (P=0.011); melanoma showed a trend (P=0.096). It was not “donor-derived bacteria newly engrafted” — it was “bacteria you originally had, of a specific kind, disappeared” that correlated with response.
6. The Identity of the Lost Bacteria — Enterocloster, Clostridium, Ruminococcus gnavus
The list of selectively lost taxa in responders contained known “bad actors” already implicated in ICI resistance. Notable examples:
- Enterocloster citroniae (formerly Clostridium citroniae)
- Enterocloster bolteae (formerly Clostridium bolteae)
- Enterocloster lavalensis
- Clostridium innocuum
- Clostridium saudiense
- Clostridium spiroforme
- Ruminococcus gnavus (recently reclassified into the genus Mediterraneibacter)
- Dialister invisus
- Sellimonas intestinalis
These taxa had been observed enriched in non-responders across earlier FMT studies (MIMIC, Baruch et al. 2021, Davar et al. 2021). FMT-LUMINate confirmed the “lost in responders” pattern using three independent detection methods (metagenomics, qPCR, culturomics).
The team further reanalyzed metagenomic data from prior FMT oncology trials and showed that the same “loss of baseline bacteria predicts response” signal was reproduced across three independent studies (pooled P=1.8 × 10⁻¹⁴). Not a FMT-LUMINate fluke — a field-wide pattern.
7. The Reverse Experiment in Mice — Lost Bacteria Abolish Antitumor Activity
So far this is observational and correlational. The crown jewel of FMT-LUMINate is the move from correlation to causation.
The team transplanted antibiotic-pretreated specific-pathogen-free (SPF) mice with stool from two NSCLC responder patients via oral gavage. The mice were then implanted subcutaneously with MCA-205 sarcoma cells and given anti-PD-1 alone or anti-PD-1 + anti-CTLA-4 intraperitoneally.
Here came the decisive intervention. From day 3 post-FMT, every 3 days, the mice were given a cocktail of the bacteria that had been lost in the responder patients — for patient R1, the four lost species (Streptococcus mutans, C. innocuum, S. parasanguinis, E. lavalensis); for patient R2, the three lost species (Enterocloster clostridioformis, S. anginosus, Clostridium tertium).
The result was clear. In mice given the lost bacteria back, ICI antitumor activity was abolished (anti-PD-1 alone P=0.028, anti-PD-1 + anti-CTLA-4 P=0.022). In control mice (responder stool only, no add-back), ICI activity was preserved.
This is direct interventional evidence that loss of baseline bacteria is necessary for FMT + ICI to work. From observational correlation to causal demonstration — a high-quality piece of mechanistic biology.
8. Why Are These Bad Bacteria Bad? — Tryptophan and the Kynurenine Pathway
With the bacterial dynamics established, the next question is “how / through what?” What did the lost bad bacteria do to block ICI?
The team profiled patient plasma metabolomics (targeted and untargeted) longitudinally. The signal that emerged was disturbance of the tryptophan metabolism pathway.
In non-responders, kynurenic acid and quinolinic acid significantly increased over time after FMT (P<0.001). Both are metabolites of the essential amino acid tryptophan flowing through the kynurenine pathway.
The kynurenine pathway is rate-limited by IDO (indoleamine 2,3-dioxygenase). Its products act through AhR (aryl hydrocarbon receptor) to expand regulatory T-cells (Tregs) and suppress CD8⁺ T-cell activation — a well-known immunosuppressive axis. Indeed, IDO inhibitors (epacadostat etc.) were tested by Merck in the KEYNOTE-252 melanoma trial in 2017–18, expected to enhance ICI but failing to improve PFS (Long et al., Lancet Oncol 2019), and the program was abandoned.
FMT-LUMINate presents a new perspective: this immunosuppressive pathway may have its source upstream in gut bacterial activity. Bad bacteria present → tryptophan over-routed to kynurenine pathway → systemic immunosuppression → ICI fails. FMT removes bad bacteria → tryptophan metabolism normalizes → suppression released → ICI works.
Indeed, in responder fecal metabolomics, tryptophan-pathway activity declined over time. Specific bacterial species were responsible for this metabolic flux, and removing them shut down the metabolic engine. Bacterial function and clinical outcome were linked at the metabolic level.
9. Immune Correlates — CD8⁺ T-Cell and Regulatory T-Cell Balance
The final mechanistic layer is host immunity. The team profiled peripheral blood mononuclear cells (PBMCs) by high-throughput flow cytometry plus plasma cytokine panels.
Comparing patients with “high subtraction” (large baseline-bacteria loss) versus “low subtraction”:
- High-subtraction patients: increased frequency of CD69⁺CD8⁺ T-cells (activation marker positive); decreased CD127low CD25high CD4⁺ regulatory T-cells (Tregs).
- Low-subtraction patients: no such changes; immunosuppressive profile maintained.
- Patients with high quinolinic acid had reduced PD-1⁺CD8⁺ T-cells and PD-1⁺CD45RA⁻CCR7⁻ effector memory CD8⁺ T-cells, and elevated regulatory T-cells.
Circulating inflammatory cytokines also shifted: high-subtraction patients showed increased IFNγ, CXCL9, CXCL13, CCL20, CD8A, CD4, and CD28 (a Th1, antitumor-favoring shift); low-subtraction patients had no major change.
So the chain — bad bacteria loss → tryptophan-metabolism normalization → IDO/AhR axis release → fewer Tregs and more activated CD8⁺ T-cells → antitumor immunity unleashed — was observed dynamically in patient blood.
10. The End of “Addition” LBPs, the Beginning of “Subtraction” Interventions
The industry implications are large.
Drug companies have spent the past decade on a long, costly path: pick good bacteria, culture them, formulate as a consortium, encapsulate, get regulatory approval. Seres Therapeutics’ VOWST (SER-109) followed this for C. difficile first, with cancer applications next. Vedanta Biosciences’ VE800 follows the same. Exeliom Biosciences’ EXL01 (F. prausnitzii-based) too.
FMT-LUMINate’s discovery shakes the foundations of all of these. Maybe “addition” LBPs work because the added good bacteria competitively exclude the bad ones — i.e., subtraction is the actual end state, addition just a mechanism to get there. If so, more direct subtraction strategies might be the real target.
Realistic options for subtraction-based intervention:
- Targeted phage therapy: bacteriophage cocktails that lyse specific deleterious species. Locus Biosciences (CRISPR-Cas3-engineered phages) in the US, Eligo Bioscience in France. C. difficile and E. coli-targeting programs are in Phase 1/2; Enterocloster-targeting next.
- Narrow-spectrum antibiotics: classic broad-spectrum antibiotics destroy entire microbiomes and worsen ICI; new agents that act on specific genera/species could be “bad-bacteria sniper rifles.” Even oral vancomycin re-evaluation is on the table.
- Competitive-exclusion consortium LBPs: blending “addition” and “subtraction.” The consortium occupies host gut niches such that bad bacteria are out-competed. Vedanta’s newer programs fit this template.
- Diet and prebiotic interventions: indirectly shift composition by feeding/starving specific taxa. Least invasive when it works, but variability is high.
Commercial timelines: phage and consortium LBPs will read out from Phase 1/2 in 2026–2027. Volume 3 covers commercialization and regulation.
11. Caveats and Limitations
The FMT-LUMINate analysis is compelling, but caveats remain.
First, the mouse experiment used a single tumor model. MCA-205 (methylcholanthrene-induced fibrosarcoma) is a classic ICI-responsive model but does not fully represent the heterogeneity of human NSCLC and melanoma biology. Whether tumor heterogeneity affects the result needs further validation.
Second, the bad-bacteria list will keep expanding. The taxa identified — Enterocloster citroniae, C. innocuum, etc. — are leading candidates, but not the complete list. Inter-individual, inter-tumor, and inter-ICI-backbone variation requires further characterization.
Third, the shift toward “function” over “species.” The real intervention target may be the metabolites bacteria produce (tryptophan products, secondary bile acids, short-chain fatty acids) rather than the bacterial species themselves. Research is deepening from species-based screening to function-based.
Summary
- FMT-LUMINate’s mechanistic analysis directly refutes the “move good bacteria from donor and add” model that has dominated FMT/LBP for a decade.
- α-diversity, β-diversity, and donor-derived-strain engraftment all showed no difference between responders and non-responders. The decisive difference was “how much of one’s own baseline bacteria was lost.”
- Lost taxa included Enterocloster citroniae, C. innocuum, E. lavalensis, R. gnavus — known ICI-resistance “bad-actor” candidates. Confirmed by three methods, reproduced across prior FMT trials.
- Reverse experiment in antibiotic-pretreated mice: reintroduction of the lost bacteria abolished antitumor activity (P=0.028, 0.022). Causality demonstrated by intervention.
- Mechanism: tryptophan-pathway disturbance → IDO/AhR activation → Treg expansion + CD8⁺ T-cell suppression → ICI resistance. FMT removes the bad bacteria, breaking the chain.
- Implications: FMT/LBP design is shifting from “addition” to “subtraction.” Phage therapy, narrow-spectrum antibiotics, competitive-exclusion consortium LBPs, and dietary interventions are the next-generation modalities.
My Thoughts and Outlook
The most striking intellectual moment of the FMT-LUMINate analysis is this: when data is collected carefully and at scale, a decade-long dominant hypothesis can be overturned by a single paper. The “lead actors” — Bacteroides, Akkermansia, F. prausnitzii — may have been “supporting actors who out-compete the villains.” Watching this field as a researcher and industry observer, my read is that this is the start of a paradigm shift for “cancer immunotherapy × gut microbiome.”
Three implications for the global research and industry community stand out. First, the locus of pharmaceutical innovation is moving one layer upstream — from the tumor-immunity interface to the systemic metabolic-immune environment. Programs at the Broad Institute, MIT-Harvard, Stanford ChEM-H, Institut Pasteur, and the Francis Crick Institute that connect microbiome biology to immunotherapy are well positioned to lead the next wave.
Second, “subtraction” modalities — particularly engineered phage therapies (Locus Biosciences’ CRISPR-Cas3 program in the US, Eligo Bioscience in France) and narrow-spectrum antibiotics — will compete head-on with the established “addition” LBP players (Seres, Vedanta, Exeliom). Investors with conviction in this paradigm shift have a multi-year window to back the right teams before the data catches up to consensus.
Third, existing pharmacology assets targeting the tryptophan–kynurenine axis (IDO inhibitor re-evaluation, AhR modulators) may resurface in microbiome-modulation contexts. The 2017–18 epacadostat story may be rewritten in a new chapter titled “combined with microbiome modulation” — turning a high-profile clinical failure into a re-positioning opportunity.
Volume 3 will cover commercialization, regulation, and partnership strategies for these next-generation modalities. From FMT, to rationally designed consortia, to subtraction interventions — we will read this 10-year story structurally.
Coming Next
The final volume dissects the other two trials — PERFORM and TACITO (both metastatic renal cell carcinoma, Nature Medicine April 2026). Healthy donors (PERFORM) vs ICI complete-responder donors (TACITO); anti-PD-1+anti-CTLA-4 backbone (PERFORM) vs anti-PD-1+axitinib backbone (TACITO) — same cancer, different donor-and-ICI combinations producing different results. We will trace the cross-trial Segatella copri toxicity-driver problem, the donor-screening implementation challenges, and the commercial frontier (Seres, Vedanta, Exeliom, Locus, Eligo) — closing the implementation-intelligence arc of the series.
Edited by the Morningglorysciences team.
Related Articles
- The Day Three Major FMT × Immunotherapy Trials Landed Together in Nature Medicine — Reshaping Cancer Immunotherapy with the Gut Microbiome | Making Cancer Immunotherapy Work with FMT, Vol. 1
- PERFORM vs TACITO, the Segatella copri Problem, and the Commercial Frontier — Three Crossroads Facing FMT’s Clinical Translation | Making Cancer Immunotherapy Work with FMT, Vol. 3 (Final)


Comments