Key Takeaways
- On April 23, 2026, Science published a landmark paper (Ciucci et al., Vol. 392, Issue 6796, DOI: 10.1126/science.ads9412) that finally provides a mechanistic answer to a long-standing epidemiological observation: why does the heart, one of the most heavily perfused organs, almost never develop cancer?
- The team combined three complementary experimental systems — an in vivo genetic mouse model, a heterotopic heart-transplantation model that surgically unloads the heart, and engineered heart tissues in which mechanical load can be tuned at will — to demonstrate that cardiac mechanical load directly inhibits cancer cell proliferation. Removing the load allowed lung adenocarcinoma, colon carcinoma, and melanoma to all grow within the myocardium.
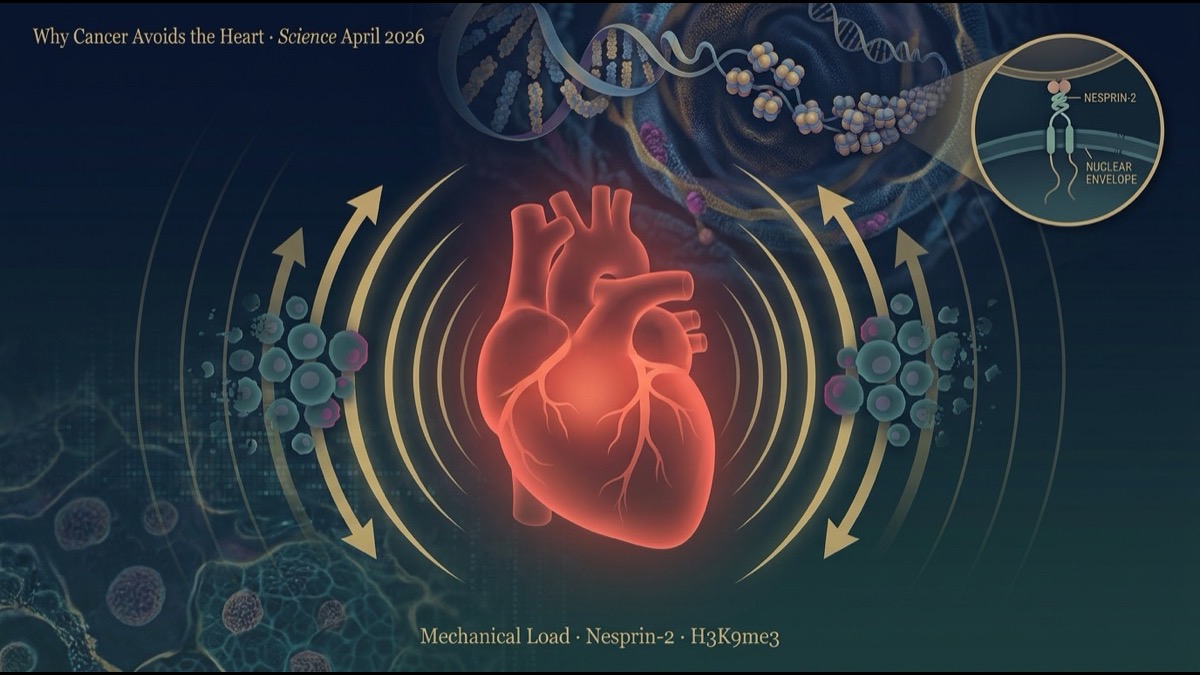
- The mechanistic core is Nesprin-2, a nuclear-envelope protein. Nesprin-2 transmits mechanical force from the cytoskeleton into the nucleus, increases histone methylation (especially H3K9 trimethylation), tightens chromatin, and reduces accessibility at proliferation-related loci. Knocking down Nesprin-2 in lung-cancer cells let them form large tumours even within mechanically loaded hearts.
- The discovery opens a new therapeutic axis — “mechanical-stimulation therapy” for cancer — and simultaneously explains why cardiac regeneration is so limited (the same mechanism stops both healthy proliferation and oncogenic proliferation). The paper sits at a unique intersection of two huge clinical fields: oncology and regenerative medicine.
Introduction — A Question Everyone Asked, Yet No One Answered
“Cancer is more common in well-perfused tissues” — this is a heuristic taught in medical school. The brain, the liver, the lungs, the kidneys: organs with rich blood supply have more primary cancers and metastases. Yet one of the most heavily perfused organs in the body — the heart — almost never develops cancer.
The autopsy incidence of primary cardiac tumours is roughly 0.001-0.03%, with the majority being benign cardiac myxomas. Malignant primary cardiac tumours (cardiac angiosarcoma, rhabdomyosarcoma, etc.) are exceedingly rare. Cardiac metastases are also strikingly less common than the heart’s share of the cardiac output would predict. In other words, the heart is the organ that “receives circulating cancer cells but does not let them settle.”
This observation has been known for decades. Three hypotheses have circulated. First: “cardiomyocytes exit the cell cycle after birth, so the surrounding microenvironment may also lack the proliferation-permissive cues cancer needs.” Second: “the dense myofibre architecture of the myocardium physically blocks tumour invasion.” Third: “cardiac-specific resident macrophages and immune populations prevent a tumour-permissive microenvironment from forming.” None of these had a rigorous molecular mechanism.
In April 2026, Ciucci et al. (International Centre for Genetic Engineering and Biotechnology, Trieste) published in Science a paper proposing a fourth, and the most experimentally validated, hypothesis: the heart suppresses cancer cell proliferation through continuous mechanical load.
This article walks through the paper’s strategy, results, mechanism, and therapeutic potential, closing with structural implications for the global research and industry community.
Main Body
1. How Rare Is Cardiac Cancer? — The Epidemiology
Before the science, a brief reminder of the numbers.
Primary cardiac tumours are found at autopsy at roughly 0.001-0.03%. The American Heart Association reports that cardiac myxomas constitute about 50% of primary cardiac tumours; the remainder are diverse benign tumours, with malignancies accounting for 10-25% (J. Am. Heart Assoc. 2020;9:e016032).
Metastatic cardiac tumours occur 20-40× more often than primary, but compared with metastases to other organs at comparable perfusion levels, they remain remarkably uncommon — autopsy detection rates are 1-15% (BMC Cancer 2018;18:202). The most likely cancers to metastasise to the heart are lung cancer (especially small cell), melanoma, lymphoma, breast cancer, and gastrointestinal cancers.
“The heart receives a comparable blood flow to many cancer-prone organs — yet cancer cells fail to colonise it.” Answering this question at a molecular level is the achievement of the present paper.
2. Lineage of Hypotheses and Open Problems
Hypotheses for cardiac cancer resistance have evolved over the past 20 years:
- Cell-cycle arrest hypothesis: Cardiomyocytes exit the cell cycle shortly after birth and remain mostly post-mitotic. The non-proliferative environment may also fail to supply proliferation cues to cancer cells.
- Anatomical-barrier hypothesis: Dense myofibre architecture and special interstitial structures physically restrict cancer cell invasion and colonisation.
- Immunological-feature hypothesis: Cardiac resident macrophages and specialised T cell subsets prevent a tumour-tolerant niche from forming.
- Metabolic-environment hypothesis: The myocardium relies primarily on fatty acid oxidation, failing to provide the glycolytic milieu (Warburg effect) that cancer cells favour.
Each contains some truth. But the mechanical-load framework — the fourth hypothesis — emerged from cardiac regenerative-medicine research as a side observation. It had been noted that one factor halting cardiomyocyte proliferation after birth is the constant “contraction-relaxation cycle.” Ciucci et al. asked whether the same mechanical environment might also block cancer cell proliferation.
3. Research Strategy — Three Experimental Systems
The paper’s strength comes from layering three complementary systems to establish causality.
System 1: A Genetic Mouse Cancer Model
Cre-mediated recombination drives K-Ras mutation overexpression and p53 deletion system-wide. Despite comparable recombination efficiency in heart, liver, and skeletal muscle, multiple cancers arose at various sites — but not a single tumour formed in the heart. The heart’s resistance to oncogenic events was confirmed in a clean genetic model.
System 2: Heterotopic Heart Transplantation Removes Mechanical Load
The aorta and pulmonary artery of a donor heart were surgically connected to the recipient mouse’s carotid artery and external jugular vein. This preserves perfusion but eliminates left-ventricular pressure-volume load — a unique system for selective mechanical unloading. Lung adenocarcinoma, colon carcinoma, and melanoma cells, when injected into unloaded hearts, all proliferated within the myocardium and formed tumours. The same cells injected into mechanically loaded hearts did not proliferate.
System 3: Engineered Heart Tissues
iPSC-derived cardiomyocytes were used to construct in vitro engineered heart tissues (EHTs) in bioreactors that allow load to be applied or removed at will. Loaded conditions suppressed cancer cell proliferation; unloaded conditions promoted it. The mechanical effect was independent of other myocardial features (extracellular matrix composition, cytokines, oxygenation).
This three-tiered design provides both observational and causal data, with each system covering the others’ limitations.
4. Result 1 — Loaded vs Unloaded: Suppression vs Tumour Formation
All three systems converged on a single conclusion: “Mechanical load suppresses cancer cell proliferation.” Lung adenocarcinoma, colon carcinoma, and melanoma cells injected into unloaded hearts all formed visible tumours within 3-4 weeks. The same cell numbers in normally loaded hearts produced no tumour.
Notably, the effect was genetic-context-independent. Lung adenocarcinoma (KRAS-driven), colon cancer (APC-loss-driven), and melanoma (BRAF-mutation-driven) — three genetically distinct cancer types — all showed the same pattern. Mechanical load thus acts through a universal mechanism that does not depend on the cancer cell’s genetic state.
5. Result 2 — Spatial Transcriptomics on Human Cardiac Metastases
Mouse models alone cannot establish human relevance. The team performed spatial transcriptomics on rare human cardiac metastases (11 cases with paired cardiac and extracardiac metastases from the same patient).
The result was striking: regardless of the primary cancer’s origin, all cardiac metastases shared a common transcriptional profile. Cancer cells that did manage to colonise the heart had been reshaped not by their primary tumour’s identity but by their “adaptation to the cardiac microenvironment.”
Among the most up-regulated genes in cardiac metastases were histone demethylases. Cardiac metastases showed reduced H3K9 trimethylation (H3K9me3) — a major repressive histone mark — and reduced chromatin compaction. The rare cancer cells that survived the cardiac microenvironment had actively rewritten their chromatin into an “open” state to escape the suppressive milieu.
6. The Mechanism — Nesprin-2 Is the Mechanosensor
Mouse models and human transcriptomics converged on histone-methylation- and chromatin-mediated proliferation control. The next question: “How does mechanical force reach the nucleus?”
The team focused on Nesprin-2 (encoded by SYNE2), a nuclear-envelope protein. Nesprin-2 forms part of the LINC (Linker of Nucleoskeleton and Cytoskeleton) complex and is known to transmit mechanical forces from the cytoskeleton to the nuclear membrane and lamina.
The functional knock-down result was dramatic: knocking down Nesprin-2 in lung cancer cells with shRNA, followed by implantation in mechanically loaded hearts, allowed the cells to form large tumours. Nesprin-2 was thus shown by a loss-of-function experiment to be a central mechanosensor.
The molecular cascade flows as follows:
- Cardiomyocyte contraction-relaxation generates mechanical forces transmitted via the extracellular matrix to engrafted cancer cells.
- The cell’s cytoskeleton (actin, microtubules) is mechanically deformed.
- Nesprin-2 functions as the bridge between the cytoskeleton and the nuclear envelope, transmitting force into the nucleus.
- Inside the nucleus: histone methyltransferase activity is modulated, particularly increasing H3K9 methylation.
- Chromatin condenses; access to proliferation-related loci (cell cycle, DNA replication, glycolysis) is reduced at promoters and enhancers.
- Result: cell proliferation halts.
Crucially, this is the same pathway by which cardiomyocytes themselves stop proliferating after birth. The reason the heart cannot regenerate (and thus the reason cancer cannot grow there) rests on a single mechano-biological principle.
7. Therapeutic Application — A “Mechanical-Stimulation Therapy” Frontier
The clinical implications are profound. The paper opens a new question: can we deploy mechanical load as a cancer therapy?
Concrete possibilities under discussion:
- Extracorporeal mechanical-stimulation therapy: Apply ultrasound, low-intensity pulsed electromagnetic fields, or vibrational stimuli to tumours to recreate mechanical load artificially. Devices analogous to those used for bone-density treatment could be adapted.
- Pharmacological activation of the Nesprin-2 pathway: Small-molecule drugs that activate Nesprin-2 without mechanical force, chemically reproducing chromatin compaction in cancer cells.
- Histone-methyltransferase activators: Activating SUV39H1, SETDB1, G9a, and other H3K9-methylation enzymes to rebuild repressive chromatin in cancer cells.
- Surgical-bed mechanical patches: Mechanical-stimulation patches (analogous to compression therapy) applied to post-surgical tumour beds to suppress residual cancer cell proliferation.
These remain conceptual and require preclinical validation, but the paper’s significance is that it has, in a reproducible system, opened a new therapeutic axis: mechanical force as an antitumour modality.
8. Caveats and Limitations
The paper is methodologically strong, but caveats remain.
First, generalisability beyond the heart. The heart is the body’s most continuously, regularly, and powerfully mechanically loaded organ. Whether the same mechanism applies to bone (dynamic loading from movement), skeletal muscle (contractile load), or uterus (stretch during pregnancy) requires separate testing.
Second, sample size of human data. Cardiac metastases are rare, and the study used 11 cases. Larger transcriptomic and spatial-omics studies are needed to confirm the mechanism in humans.
Third, Nesprin-2 alone or with other mechanosensors? The LINC complex has other members (Nesprin-1, -3, -4, SUN1, SUN2). Their relative contributions and redundancy are open questions.
Fourth, the long road to clinical translation. “Mechanical stimulation as cancer therapy” is conceptually attractive, but: (a) precisely targeting tumours with mechanical force, (b) avoiding side effects on healthy tissue, (c) reaching disseminated metastatic disease via mechanical means — all remain unsolved. Realistic clinical translation is on a >10-year horizon.
Summary
- Ciucci et al. in Science (April 2026) explained why the heart almost never develops cancer through mechanical-load-dependent chromatin regulation.
- Three experimental systems — genetic mouse model + heterotopic transplantation unloading + engineered heart tissues — established that mechanical load suppresses cancer cell proliferation. Removing load allowed lung adenocarcinoma, colon carcinoma, and melanoma all to grow in the myocardium.
- Spatial transcriptomics on human cardiac metastases revealed a common signature regardless of primary cancer type: up-regulation of histone demethylases and reduced H3K9me3.
- The molecular cascade is Nesprin-2 → histone methylation → chromatin compaction → reduced accessibility at proliferation loci. Nesprin-2 knock-down directly demonstrated causality.
- Therapeutic applications: mechanical-stimulation therapy, Nesprin-2 pathway activators, H3K9 methyltransferase activators — a new therapeutic axis.
- Limitations: generalisability to other mechanically loaded organs (bone, skeletal muscle, uterus), human sample size, contributions of other LINC complex members, and the long timeline to clinical translation.
My Thoughts and Outlook
The deeper value of this paper is that it links two large research fields — cancer research and regenerative medicine — through a single mechano-biological principle. The reason cardiomyocytes cannot regenerate (mechanical-load-dependent chromatin compaction halts proliferation) and the reason the heart resists cancer are two sides of the same coin. From the same molecular targets (Nesprin-2 / H3K9 methylation), one can derive opposing therapeutic strategies: “to promote regeneration, release mechanical load” and “to suppress cancer, apply mechanical load.”
Three structural implications for the global research and industry community. First, mechanobiology becomes a mainstream therapeutic area. Centres including the Wyss Institute (Harvard), the Mechanobiology Institute Singapore, and groups at Imperial College London and ETH Zurich are well positioned to extend Ciucci et al.’s findings into preclinical drug discovery and device development. Second, non-invasive mechanical-stimulation devices represent an underbuilt therapeutic category. Ultrasound-based focused therapy companies (Insightec, Profound Medical) and low-intensity pulsed electromagnetic-field developers may find a new oncology indication path. Third, iPSC-based engineered heart tissues become not just regenerative-medicine tools but also platforms for cancer drug discovery. Integrated platforms — combining iPSC differentiation, bioreactor-based mechanical loading, and high-content imaging — are a competitive advantage that will determine which institutions lead the next 5-10 years of mechanobiology-oncology research.
2026 is the year AI is rapidly commoditising knowledge work. That makes the value of physical-tissue-based therapeutic interventions go up, not down. Mechanical-stimulation therapy is precisely such a “value-rises-in-the-AI-age” field — requiring physical hardware, regulated medical devices, and clinician-administered intervention. A single paper has opened a frontier that may take a decade to traverse, but the destination is significant.
Edited by the Morningglorysciences team.
Related Articles
- Where Will Nuclear Medicine Be in the Next Decade? Ac-225, Pb-212, and the Next-Generation RI Pipeline Map | Vol.3 (Final)
- Mapping Nuclear Medicine Competitors: Lantheus, Bayer, Telix and the 3 Turning Points Ahead | Vol.2
- Where Is Nuclear Medicine Heading? Pluvicto’s Success and the Vertical Integration Imperative | Nuclear Medicine Vol.1
- Does Whole-Genome Sequencing Truly Help in Solid-Tumor Clinical Care? — Real-World Clinical Utility Validation | Nature Medicine April 2026
- Where Is CAR-T Heading? 3 Commercialization Paths (Autologous/Allogeneic/In Vivo) and the Next-Decade Optimal Mix | Vol.3 (Final)


Comments