
要点まとめ
- 2026年5月14日号 Cell 誌に、がん発生の「窓」を空間トランスクリプトミクスで解明した重要論文(Reyes et al., Memorial Sloan Kettering Cancer Center, DOI: 10.1016/j.cell.2026.03.032)が掲載されました。膵管腺癌(PDAC)モデルで、良性病変から悪性がんへの転換点が、希少な「progenitor-like 細胞集団」とそれが組織する「自己強化型ニッチ」で起こることを示した研究です。
- 研究グループは KrasG12D マウス+自然 p53 LOH モデル(KPLOH)を使い、単細胞 RNA-seq+空間トランスクリプトミクス+多重免疫染色を統合。p53 活性または KRAS シグナル抑制で progenitor niche が崩壊し、がん進展が遅延 することを示しました。
- 機序の核心は 「がん駆動変異とがん抑制プログラムの両方が同じ progenitor 細胞内で同時に活性化している」という所見。p53、CDKN2A、SMAD4 という3つの主要ながん抑制プログラムがすべて progenitor 細胞で稼働し、その崩壊が悪性化の引き金となります。
- 臨床的含意:「がんになる前に介入する」新しい時間軸が開きました。Progenitor 細胞集団を標的にする介入は、確立されたがんを治療するより遥かに有効である可能性。膵がん早期発見・予防医療への応用、KRAS 阻害薬(Sotorasib, Adagrasib, RMC-6236)の前がん病変への適応拡大の道が見えます。
序論——「クローン進化」と「がん幹細胞」の先へ
がんはどう始まるのか。この問いに対する20世紀的な答えは「クローン進化(Nowell, 1976)」でした。突然変異が蓄積し、選択的に有利なクローンが拡大し、最終的に悪性化する、という直線的モデルです。21世紀に入って「がん幹細胞仮説(Reya et al., 2001)」が加わり、特定の幹細胞様集団ががん発生の「種」として機能する、という空間的概念が導入されました。
しかし、過去10年の単細胞解析・空間オミクス・系統追跡技術の急速な発展は、これら旧来モデルの単純化に挑戦しています。例えば「正常組織にもがん駆動変異を持つクローンが多数潜伏している」(Martincorena et al., Science 2018; Yokoyama et al., Nature 2019)という発見は、「変異の蓄積だけではがんにならない」ことを明示しました。何かが追加で必要なのです。
2026年5月号 Cell 誌の Reyes et al. は、その「何か」が 「progenitor-like 細胞集団とそれが組織する自己強化型ニッチ」 である、という具体的回答を提示します。膵管腺癌(PDAC)という、KRAS変異が95%を占める典型的「変異駆動型がん」を舞台に、変異だけでは説明できない「組織レベルの転換点」を浮き彫りにしました。
本記事は、本論文の研究戦略・主要結果・機序・治療応用可能性を解説し、後半で日本の研究・産業への含意を整理します。
本論
1. 膵管腺癌(PDAC)はなぜ良いモデルか
本論文が PDAC モデルを採用したのは戦略的な選択です。PDAC は以下の特徴で「がん起源研究の理想モデル」を成しています。
- KRAS 変異が95%以上 で、駆動変異が単純化されている
- 段階的進展 が比較的明瞭(PanIN-1A → PanIN-1B → PanIN-2 → PanIN-3 → 浸潤癌)
- 主要がん抑制因子 が3つに集約(TP53、CDKN2A、SMAD4)
- マウス遺伝モデル が確立(KC、KPC、KPLOH)
- 5年生存率10%未満 という臨床的緊急性
研究グループは KPLOH モデル(KrasG12D, Trp53flox/+, Ptf1a-Cre + 蛍光レポーター)を使い、自然発生する p53 ヘテロ接合性消失(LOH)を蛍光標識で追跡可能にしました。これにより、p53 LOH 直後の細胞を1個1個サンプリングできます。膵がんが顕在化する前の「窓」を捕捉する設計です。
2. 単細胞 RNA-seq が見つけた稀な「progenitor-like 集団」
scRNA-seq で前がん段階の KrasG12D 上皮細胞を解析すると、複数の前がん細胞状態が現れました。腺房-管移行(ADM, acinar-to-ductal metaplasia)、神経内分泌様、tuft 細胞様、増殖性、胃様(PanIN/古典的 PDAC サブタイプ)――いずれも既知の状態です。
その中で、研究グループは 稀な集団 に注目します。Nes(ネスチン)、Msn(モエシン)、Hmga2、Hmga1、Vim という間葉系・幹細胞性マーカーを発現する progenitor-like 状態 です。
注目すべきは、この progenitor-like 集団が 「PDAC(=最終悪性状態)に転写プロファイルとして最も近い」ことでした。前がん段階のあらゆる細胞状態の中で、progenitor-like 細胞が「悪性への跳躍距離」を最も短く保っている――つまり、悪性化の有力な「中間状態」として浮上したのです。
3. 「がん駆動」と「がん抑制」が同じ細胞で同時稼働する
論文の最も衝撃的な発見は、この progenitor-like 細胞内で起きていた「綱引き」でした。
研究グループは、scRNA-seq データを使って p53、CDKN2A、SMAD4 というPDAC の3大がん抑制プログラムの活性を細胞集団ごとに比較。すると、これら3プログラムすべてが progenitor-like 細胞で最も強く稼働 していたのです。
具体的には:
- p53 標的遺伝子(Cdkn1a, Bax, Bbc3, Mgmt, Mdm2 等):progenitor-like 細胞で最高発現
- Cdkn2a(p16INK4A、p19ARF):progenitor-like 細胞で発現上昇、p16/p19 両アイソフォーム使用
- SMAD4 / TGF-β 経路標的遺伝子:progenitor-like 細胞で活性化
同時に、がん駆動シグナル(KRAS下流の Fosl1、解糖系/Warburg、上皮間葉転換 [EMT])も同じ細胞で活性化していました。つまり、この細胞集団は「がんになる」ベクトルと「がんを抑える」ベクトルの両方を同時に発動している、極めて不安定な状態だったのです。
これは古典的な 「腫瘍化シグナル誘導性老化(oncogene-induced senescence, OIS)」 概念と整合します。RAS 経路の異常活性化が p53/p16INK4A 依存の細胞老化を誘導し、がん細胞の増殖を抑える、という30年来の理論が、空間的・細胞集団特異的に再確認されたのです。
4. Spatial Transcriptomics が見せた「ニッチ」の正体
scRNA-seq だけでは「細胞同士の空間関係」は分かりません。研究グループは Xenium In Situ プラットフォーム(10× Genomics)で空間トランスクリプトミクスを実施しました。480遺伝子パネルで350万細胞以上を解析。
結果、progenitor-like 細胞は組織内で 偶発的に分散している のではなく、特定の細胞種を周囲に集めた「ニッチ」を形成 していました。
具体的には:
- Itgax+ TAM(腫瘍関連マクロファージ):免疫抑制機能を持つ亜集団が progenitor 周囲に集積
- Tnc+ myCAF(活性化筋線維芽細胞):細胞外マトリクス(ECM)を産生し、組織硬化に寄与
- TGF-β 関連シグナル:上皮 → 線維芽細胞 → 免疫細胞の三者間で活発化
- 創傷治癒応答プログラム:複数の細胞種で並行活性化
このニッチは 「PDAC が確立した後の腫瘍微小環境」と非常によく似た構造 でした。つまり、組織がまだ「がん」と診断されない前段階で、すでに「がんと同じ社会構造」が小規模に組み上がり始めている、という驚くべき所見です。
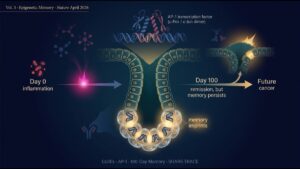
5. 自己強化型ニッチ——不可逆的な転換が起こる仕組み
このニッチの本質は 「自己強化ループ」 にあります。研究グループは ligand-receptor pair 解析と niche pseudotime(ニッチの時間進行を空間データから推定する手法)を用いて、ニッチが時間発展する様子を再構成しました。
動的な姿は以下の通り:
- Step 1:KRAS 異常活性化により稀な細胞が progenitor-like 状態へ転移
- Step 2:progenitor-like 細胞が周囲に PDGF、TGF-β、IL-6 等のサイトカインを分泌
- Step 3:これらのサイトカインが myCAF・TAM を集積、活性化、極性化(M2型・免疫抑制型へ)
- Step 4:myCAF が ECM(Tnc, Postn)と TGF-β を産生し、上皮細胞をさらに progenitor-like 状態へ押し込む
- Step 5:TAM が Arg1 等を産生し、CD8⁺ T 細胞排除=免疫監視回避を確立
- Step 6:ニッチが拡大、より多くの上皮細胞を取り込んで悪性転換を加速
この「自己強化ループ」が確立すると、p53/CDKN2A/SMAD4 の抑制力では止められなくなり、最終的にがん抑制プログラムが脱落(変異・LOH・エピジェネティックサイレンシング)して悪性転換が確立します。
6. KRAS 阻害または p53 活性化でニッチが崩壊する
ここから論文は治療応用への橋を架けます。「もしニッチが自己強化ループなら、ループのいずれかの輪を切れば崩壊する」 という仮説を、2つの介入実験で検証しました。
介入1:KRAS 阻害
KRAS-G12D 阻害薬(Adagrasib 類似)の一過性投与で、progenitor-like 細胞集団が枯渇。ニッチを支える「中心細胞」がいなくなることで、myCAF と TAM の集積が解除され、ニッチ全体が解体されました。結果、悪性化への進展が遅延しました。
介入2:p53 活性化
p53 機能を強化する遺伝学的介入で、progenitor-like 細胞が選択的にアポトーシスまたは老化に追い込まれ、ニッチが崩壊。同様に悪性化が遅延しました。
逆に p53 抑制 を行うと、progenitor-like 細胞が拡大、上皮間葉転換が進行、免疫排除型ニッチが完全形成されて、悪性化が加速しました。
これらの実験は、「progenitor niche を標的にする介入は確立されたがんを治すより効率的に悪性化を阻止できる」という、治療学的に大きな含意を持ちます。
7. ヒト膵炎組織でも同じニッチが見える
マウスでの所見はヒトでも妥当か? 研究グループは、慢性膵炎・PDAC隣接組織67検体で多重免疫染色を実施。マウス progenitor-like 細胞のヒト相同マーカーとして KRT17 陽性 basal-like 細胞 を採用しました。
結果、ヒト慢性膵炎組織でも:
- KRT17+ 細胞の周囲に TNC+ 線維芽細胞、ARG1+ 免疫抑制マクロファージが集積
- p53、p16INK4A、CRYAB(老化マーカー)の発現亢進
- CD8a・GZMB(細胞傷害性T細胞)の排除
という、マウス progenitor niche と 瓜二つの構造 が観察されました。これは「ヒト膵がんはまだ顕在化していない段階で、既に分子的・組織学的に転換が始まっている」ことを意味します。臨床的早期介入の窓が、想定より広い可能性が示唆されます。
8. 研究の限界と注意点
強力な研究ですが限界もあります。
第一に、PDAC 特異性 vs 普遍性:「progenitor niche モデル」が他のがん(大腸がん、肺腺がん、乳がん等)にも当てはまるかは別途検証が必要。すでに肺腺がん・大腸がんで類似所見が部分的に報告されていますが、組織別の機序差は今後の研究課題です。
第二に、ヒトでの介入実験の不可能性:マウスでは KRAS 阻害・p53 活性化で「ニッチが崩壊し悪性化が遅延する」を直接証明できますが、ヒトでは実施できません。間接的証拠の積み上げが必要です。
第三に、progenitor-like 集団の同定マーカーの限界:Msn、Nes、Hmga2 等は progenitor-like 状態のマーカーですが、これらだけで臨床的に「ニッチ陽性組織」を診断するには感度・特異度が不足。今後は分子複合シグネチャによる診断キット開発が要ります。
第四に、治療の時間窓:「ニッチが完全形成される前に介入する」が理想ですが、ヒトでは膵がんが診断可能な時点でニッチは既に成熟している可能性が高い。「ニッチを後戻りさせる」治療概念の検証も別途必要です。
まとめ
- Reyes et al. Cell 2026 は、PDAC 発生過程で良悪性転換が起こる「窓」が、希少な progenitor-like 細胞集団とそれが組織する自己強化型ニッチ であることを単細胞・空間オミクス・組織学・介入実験の統合で示した。
- Progenitor-like 細胞では がん駆動シグナル(KRAS、解糖系、EMT)と がん抑制シグナル(p53、CDKN2A、SMAD4)が同時稼働、典型的な「腫瘍化シグナル誘導性老化」状態にある。
- このニッチは Itgax+ TAM、Tnc+ myCAF、TGF-β/創傷治癒経路 という多細胞種参加の構造で、PDAC 微小環境を縮小再現している。
- KRAS 阻害または p53 活性化で ニッチが崩壊し悪性化が遅延。治療学的標的としての validity が示された。
- ヒト慢性膵炎組織でもマウスと同じニッチが KRT17+ basal-like 細胞中心で観察され、臨床的早期介入の窓 が想定より広い可能性。
- 限界:PDAC 特異性 vs 普遍性、ヒトでの介入実験不可能性、診断マーカーの臨床応用ハードル、治療時間窓の現実的制約。
私の考察・展望
本論文の最大の知的貢献は、「がん発生は変異だけでなく、組織レベルの動的状態(ニッチ)によって制御される」というモデルを、空間オミクス・介入実験で確立したことです。これまで「変異の蓄積で不可避にがんになる」と考えられてきたシナリオが、「変異があってもニッチが組み上がらないとがんにならない」へと書き換えられました。これは、がん予防医療の概念を根本的に変える発見です。
日本の研究・産業にとっての示唆は3点。第一に、空間オミクス研究基盤 の活用。日本は理研IMS、東大、東京医科歯科大、京大医学部などで Xenium・MERFISH等の空間トランスクリプトミクスプラットフォームが整備されつつあります。膵がんの早期病変・PanIN 検体の二次利用で、Reyes ら の所見を日本人コホートで再現する研究は、世界に貢献できる立ち位置にあります。第二に、KRAS 阻害薬の早期介入適応拡大。Sotorasib(Amgen)・Adagrasib(Mirati)・Revolution Medicines の RMC-6236 が次々と上市・後期試験中の今、これらを「確立 PDAC への治療」だけでなく「PanIN-3 や IPMN といった前がん病変への介入」に拡張する研究設計が成り立ちます。日本の膵癌外科・内科は世界トップクラスで、こうした介入試験プロトコル設計の主体になれます。第三に、診断キット開発。「progenitor-niche 陽性組織」をピンポイントで検出する病理診断キット(多重免疫染色+AI画像解析)は、世界市場で需要が立ち上がる典型的な「研究-診断-治療」連結機会。日本の臨床診断企業(シスメックス、栄研化学)と病理AI企業(メドメイン、アイリス)の協業で展開可能です。
連載第2回では、ヒト小脳オルガノイドで髄芽腫の発生・進展を再現した別の重要論文を扱います。これも本論文と並ぶ「がん起源研究の新世代モデル」を象徴する研究です。
次回予告
連載第2回は、 Cell 誌2026年4月号に同時期掲載された 「ヒト小脳オルガノイドにおける髄芽腫の発生・進展・治療応答モデル」 論文を扱います。患者由来 iPSC から分化させた小脳オルガノイドに、臨床で観察される髄芽腫遺伝子変異を導入し、発生過程をリアルタイムで再現。本論文の progenitor niche モデルと並んで、「がん起源を実験室で再現する」新世代研究の到達点を示します。
Morningglorysciencesチームによって編集されました。


コメント