
Key Takeaways
- The May 14, 2026 issue of Cell published a landmark paper (Reyes et al., Memorial Sloan Kettering Cancer Center, DOI: 10.1016/j.cell.2026.03.032) that uses spatial transcriptomics to dissect the “window” where cancer arises. In a pancreatic ductal adenocarcinoma (PDAC) model, the team showed that the benign-to-malignant transition happens within a rare “progenitor-like cell population” and the self-reinforcing niche they assemble.
- Using a KrasG12D mouse model with spontaneous p53 LOH (KPLOH), the team integrated single-cell RNA-seq, spatial transcriptomics, and multiplex immunofluorescence. Either p53 reactivation or transient KRAS inhibition collapses the progenitor niche and delays malignant progression.
- The mechanistic core is the surprising finding that oncogenic and tumor-suppressive programs are co-activated in the same progenitor cells. p53, CDKN2A, and SMAD4 — the three major tumor-suppressor programs of PDAC — all run in progenitor-like cells, and the collapse of these programs triggers full malignancy.
- Clinical implications: a “intervene before cancer becomes cancer” time horizon. Targeting the progenitor cell population may be far more effective than treating established cancer. Possibilities include early detection / preventive medicine for pancreatic cancer, and extending the indication of KRAS inhibitors (Sotorasib, Adagrasib, RMC-6236) to premalignant lesions.
Introduction — Beyond “Clonal Evolution” and “Cancer Stem Cells”
How does cancer begin? The 20th-century answer was the clonal-evolution model (Nowell, 1976): mutations accumulate, selectively advantageous clones expand, and malignancy follows along a linear trajectory. The 21st century added the cancer-stem-cell hypothesis (Reya et al., 2001), which introduced the spatial concept that a specific stem-like population may be the “seed” of cancer initiation.
But the rapid maturation of single-cell, spatial-omics, and lineage-tracing technologies over the past decade has challenged the simplicity of these models. The discovery that “many normal tissues harbor large clones bearing canonical cancer driver mutations” (Martincorena et al., Science 2018; Yokoyama et al., Nature 2019) made it explicit: mutation accumulation alone is not enough to make cancer. Something else is required.
Reyes et al. in the May 2026 Cell issue propose a concrete answer: that “something else” is a progenitor-like cell population and the self-reinforcing niche it assembles. Using PDAC — the prototypical mutation-driven cancer (>95% KRAS-mutant) — they spotlight a tissue-level inflection point that mutations alone cannot explain.
This article walks through the paper’s strategy, principal results, mechanism, and therapeutic potential, closing with structural implications for the global research and industry community.
Main Body
1. Why PDAC Is the Right Model
The choice of PDAC is strategic. PDAC offers an almost-ideal model for cancer-origin research:
- >95% KRAS mutation — driver mutations are simplified.
- Stepwise progression is well defined (PanIN-1A → 1B → 2 → 3 → invasive carcinoma).
- Major tumor suppressors are concentrated in three: TP53, CDKN2A, SMAD4.
- Mouse models are well established: KC, KPC, KPLOH.
- Less than 10% 5-year survival — clinical urgency.
The team used the KPLOH model (KrasG12D, Trp53flox/+, Ptf1a-Cre + fluorescent reporters) to track spontaneous p53 LOH cell-by-cell. This enables sampling of cells immediately after p53 LOH — a design that captures the “window” before macroscopic pancreatic cancer.
2. Single-Cell RNA-seq Found a Rare “Progenitor-Like Population”
scRNA-seq of premalignant KrasG12D epithelial cells revealed multiple premalignant cell states: acinar-to-ductal metaplasia (ADM), neuroendocrine-like, tuft-like, proliferative, gastric-like (PanIN / classical PDAC subtype) — all known states.
Among these, the team highlighted a rare population expressing the mesenchymal/stem-like markers Nes (nestin), Msn (moesin), Hmga2, Hmga1, and Vim — the progenitor-like state.
The striking observation: this progenitor-like population was “transcriptionally closest to PDAC (the final malignant state)” among all premalignant cell states. The progenitor-like cells maintained the shortest “leap distance” to malignancy — emerging as the leading “intermediate state” for the benign-to-malignant transition.
3. “Cancer Drivers” and “Tumor Suppressors” Co-Active in the Same Cell
The paper’s most striking finding was a “tug of war” inside these progenitor-like cells.
The team compared activity of the three major PDAC tumor suppressors — p53, CDKN2A, SMAD4 — across cell populations. All three programs were most strongly active in progenitor-like cells:
- p53 target genes (Cdkn1a, Bax, Bbc3, Mgmt, Mdm2, etc.): peak expression in progenitor-like cells.
- Cdkn2a (p16INK4A, p19ARF): elevated, with both isoforms engaged.
- SMAD4 / TGF-β-pathway targets: activated.
Simultaneously, cancer-driving signals (KRAS-downstream Fosl1, glycolysis/Warburg, epithelial-mesenchymal transition) were also active in these same cells. The progenitor-like population was running both “becoming cancer” vectors and “stopping cancer” vectors at the same time — a profoundly unstable state.
This aligns with the classical concept of “oncogene-induced senescence (OIS)”: aberrant RAS activation triggers p53/p16INK4A-dependent cellular senescence that blocks cancer growth. The 30-year-old theory was reaffirmed at spatial- and population-specific resolution.
4. Spatial Transcriptomics Revealed the “Niche”
scRNA-seq alone cannot resolve “spatial relationships between cells.” The team performed spatial transcriptomics with the Xenium In Situ platform (10× Genomics), profiling >3.5 million cells with a 480-gene panel.
Progenitor-like cells in tissue were not randomly dispersed: they assembled specific surrounding cell types into a “niche”.
The niche components:
- Itgax+ TAM (tumor-associated macrophages) — immunosuppressive subsets clustering around progenitors.
- Tnc+ myCAF (activated myofibroblasts) — extracellular matrix producers, contributing to tissue stiffening.
- TGF-β-related signaling — active among epithelium, fibroblasts, and immune cells.
- Wound-healing program — co-activated across multiple cell types.
This niche structure closely mimics the tumor microenvironment of established PDAC. In other words, before tissue is even diagnosable as cancer, a “society identical to cancer’s” is already being built up at small scale — a stunning observation.
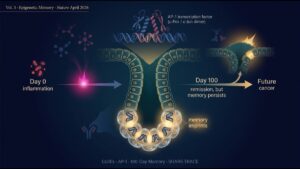
5. The Self-Reinforcing Niche — How an Irreversible Transition Occurs
The niche’s essence is its “self-reinforcing loop.” Using ligand-receptor pair analysis and “niche pseudotime” (a method to infer niche temporal evolution from spatial data), the team reconstructed dynamic niche progression:
- Step 1: Aberrant KRAS activation pushes a rare cell into the progenitor-like state.
- Step 2: Progenitor-like cells secrete PDGF, TGF-β, IL-6, and other cytokines.
- Step 3: These cytokines recruit and polarize myCAFs and TAMs (toward M2 / immunosuppressive).
- Step 4: myCAFs produce ECM (Tnc, Postn) and TGF-β, pushing more epithelial cells into the progenitor-like state.
- Step 5: TAMs produce Arg1 and other factors, excluding CD8⁺ T cells = immune escape established.
- Step 6: The niche expands, drawing in more epithelial cells and accelerating malignant transition.
Once this self-reinforcing loop is established, p53/CDKN2A/SMAD4 suppression cannot keep up. Eventually, tumor-suppressor programs are lost (mutation, LOH, epigenetic silencing), and full malignancy is locked in.
6. KRAS Inhibition or p53 Activation Collapses the Niche
The paper builds a bridge to therapeutic application: “if the niche is a self-reinforcing loop, breaking any one link should collapse it.” They tested this with two interventions.
Intervention 1: KRAS Inhibition
A transient KRAS-G12D inhibitor (Adagrasib-like) depleted the progenitor-like cell population. With the “central cells” of the niche gone, myCAF and TAM accumulation dissolved, and the entire niche fell apart. Result: malignant progression was delayed.
Intervention 2: p53 Activation
Genetic enhancement of p53 function selectively drove progenitor-like cells into apoptosis or senescence, collapsing the niche. Malignant progression similarly delayed.
Conversely, p53 suppression expanded progenitor-like cells, advanced epithelial-mesenchymal transition, and produced a fully formed immune-exclusion niche, accelerating malignancy.
These experiments carry major therapeutic implications: targeting the progenitor niche may stop malignancy more efficiently than treating established cancer.
7. The Same Niche Is Visible in Human Pancreatitis Tissue
Are mouse findings valid in humans? The team performed multiplex immunofluorescence on 67 chronic pancreatitis and PDAC-adjacent tissue cores. They used KRT17+ basal-like epithelial cells as the human ortholog of mouse progenitor-like cells.
In human chronic pancreatitis tissue:
- KRT17+ cells were surrounded by TNC+ fibroblasts and ARG1+ immunosuppressive macrophages.
- p53, p16INK4A, and CRYAB (senescence marker) were elevated.
- CD8a and GZMB (cytotoxic T cells) were excluded.
The structure was nearly identical to the mouse progenitor niche. This means human pancreatic cancer likely begins its molecular and tissue-level transition long before clinical diagnosis. The window for early intervention may be wider than presumed.
8. Limitations and Caveats
The paper is rigorous, but limitations remain.
First, PDAC-specificity vs universality. Whether the “progenitor niche model” applies to other cancers (colon, lung adenocarcinoma, breast) requires separate testing. Partial findings in lung adenocarcinoma and colon cancer have been reported, but tissue-specific differences are open questions.
Second, the impossibility of intervention experiments in humans. Mouse experiments directly demonstrated that “KRAS inhibition / p53 activation collapses the niche and delays malignancy,” but such experiments cannot be performed in humans. Indirect evidence must accumulate.
Third, marker limitations for the progenitor-like population. Msn, Nes, Hmga2 mark progenitor-like cells but lack sensitivity and specificity for clinical diagnosis. Future work needs molecular composite signatures and diagnostic kit development.
Fourth, the therapeutic time window. Intervening “before the niche fully assembles” is ideal, but in humans, the niche is likely already mature by the time pancreatic cancer becomes diagnosable. The concept of “rolling back the niche” needs separate validation.
Summary
- Reyes et al. Cell 2026 demonstrated that the benign-to-malignant transition in PDAC occurs within a rare progenitor-like cell population and the self-reinforcing niche it assembles, integrating single-cell, spatial-omics, histology, and intervention experiments.
- In progenitor-like cells, cancer-driving signals (KRAS, glycolysis, EMT) and tumor-suppressing signals (p53, CDKN2A, SMAD4) are co-active — a textbook “oncogene-induced senescence” state.
- The niche assembles Itgax+ TAMs, Tnc+ myCAFs, TGF-β / wound-healing pathways, recapitulating the PDAC microenvironment in miniature.
- KRAS inhibition or p53 activation collapses the niche and delays malignancy, validating its therapeutic relevance.
- The same niche, centered on KRT17+ basal-like cells, is observed in human chronic pancreatitis tissue, suggesting a wider window for early intervention than presumed.
- Limitations: PDAC-specificity vs universality, impossibility of human intervention experiments, clinical-translation hurdles for diagnostic markers, realistic constraints on therapeutic time windows.
My Thoughts and Outlook
The paper’s deepest contribution is establishing — through spatial omics and intervention experiments — the model that “cancer initiation is regulated not only by mutations but by tissue-level dynamic states (niches).” The previously dominant scenario, “mutation accumulation inevitably leads to cancer,” is being rewritten as “mutations alone do not produce cancer if the niche fails to assemble.” This fundamentally changes the conceptual framework for cancer prevention.
Three structural implications for the global research and industry community. First, spatial-omics platforms are becoming foundational infrastructure for translational cancer research. Centers like Memorial Sloan Kettering, the Broad Institute, the Sanger Institute, the Karolinska Institute (which hosts substantial Xenium / MERFISH installations), and Singapore’s A*STAR are well positioned to extend the Reyes et al. findings across cancer types. Second, the indication scope of KRAS inhibitors is expanding beyond established cancer. With Sotorasib (Amgen), Adagrasib (Mirati / Bristol Myers Squibb), and Revolution Medicines‘ RMC-6236 entering or in late-stage trials, the next frontier is “intervention in PanIN-3 / IPMN / other premalignant lesions.” Trial protocols for this expansion are now technically feasible. Third, diagnostic-kit development for “progenitor niche-positive tissue” is an underbuilt category. Multiplex immunofluorescence + AI image analysis kits that pinpoint KRT17+ cells with associated immunosuppressive niches represent a clinical opportunity that diagnostic specialists (Roche, Agilent, Leica Biosystems, Indica Labs) and AI pathology firms (PathAI, Paige) can pursue.
2026 is the year AI is rapidly commoditizing knowledge work. Cancer-origin research, which requires physical tissue specimens, advanced imaging hardware, and clinically integrated protocols, sits in the “AI cannot replace” zone. This is precisely why mechanistic discoveries like Reyes et al. command structural value — they create new clinical workflows, new device categories, and new therapeutic indications that AI alone cannot produce.
Volume 2 of this series turns to the Cell paper that uses human cerebellar organoids to model medulloblastoma development — another landmark of the new generation of cancer-origin research.
Coming Next
Volume 2 examines the Cell 2026 April paper “Modeling medulloblastoma pathogenesis and treatment in human cerebellar organoids.” Patient-derived iPSC-differentiated cerebellar organoids are engineered with clinically observed medulloblastoma mutations to recapitulate development in real time. Together with the progenitor-niche model in Volume 1, this represents the cutting edge of “reproducing cancer origin in the laboratory.”
Edited by the Morningglorysciences team.


Comments