Key Takeaways
- The April 2026 issue of Cell published a landmark paper that uses human iPSC-derived cerebellar organoids to recapitulate medulloblastoma development, progression, and therapeutic response in real time. Together with the progenitor-niche paper from Volume 1, this represents a generational shift in cancer-origin research.
- Medulloblastoma is the most common malignant brain tumor in children. It is classified into four molecular subtypes (WNT, SHH, Group 3, Group 4) with vastly different survival rates and treatment responses. The team introduced subtype-defining mutations (CTNNB1, PTCH1, MYC, OTX2, etc.) into healthy donor iPSCs by CRISPR and reproduced full developmental trajectories in cerebellar organoids.
- Two principal findings. First, the cell of origin for each subtype was directly tied to specific developmental timing and progenitor populations in the human cerebellum. Second, subtype-specific drug targets (SMO/GLI for SHH; BET/mTOR for Group 3) were validated for response in organoids.
- Clinical implications: an era of “per-patient medulloblastoma organoids” for treatment selection. Vismodegib (SHH/Hh inhibitor), BET inhibitors, and CAR-T combinations can be optimized via pre-treatment organoid assays — predicting response before exposing patients.
Introduction — How Do You Study a Cancer That Mice Cannot Reproduce?
Medulloblastoma occupies a special place in cancer research. The most common malignant pediatric brain tumor, it peaks in onset around age 5, arising in the cerebellum — an anatomically inaccessible site. Five-year survival has improved to about 75%, but most survivors carry radiation-induced cognitive impairment — a quintessential “treated, but scarred” cancer.
For 30 years, medulloblastoma research has had a fundamental challenge: mouse models cannot fully reproduce human medulloblastoma. Mouse and human cerebellar development, cell-type composition, and gene expression diverge substantially, particularly in the granule neuron precursor (GNP) population specific to cerebellum. Mouse models for SHH-subtype medulloblastoma (e.g., heterozygous Ptch knockouts) reproduced part of the disease, but Group 3 and Group 4 — major subtypes by frequency — have no faithful mouse models. This is a research wall.
The April 2026 Cell paper broke through this wall by introducing each subtype’s driver mutations into human iPSC-derived cerebellar organoids by CRISPR. The work is more than “a new model” — it solves both the cell of origin and the therapeutic response for each subtype, updating the medulloblastoma research paradigm.
This article walks through the paper’s strategy, principal results, and clinical-application implications, with structural reflections for the global research and industry community.
Main Body
1. The Clinical Significance of the Four Medulloblastoma Subtypes
Quick recap of the four molecular subtypes (Taylor et al., Acta Neuropathol 2010):
| Subtype | Frequency | Driver mutations | 5-yr survival | Cell of origin |
|---|---|---|---|---|
| WNT | 10% | CTNNB1 mutations | ~95% | Lower rhombic lip |
| SHH | 30% | PTCH1, SMO mutations; GLI2 amp | 60-80% | External granule layer GNP |
| Group 3 | 25% | MYC amplification | 40-60% | Unknown (candidates: granule precursors, Nestin+ stem cells) |
| Group 4 | 35% | OTX2/MYCN abnormalities, KDM6A, etc. | 60-75% | Unknown (candidates: unipolar brush cell, posterior rhombic lip precursor) |
By subtype:
- Prognosis varies dramatically: WNT best, Group 3 worst.
- Standard therapy responses differ: chemotherapy, radiation, SHH inhibitors all show subtype-dependent efficacy.
- Targeted-therapy candidates differ: SMO inhibitors only for SHH; MYC-targeting strategies particularly relevant to Group 3.
Confirming subtype is now standard, with subtype-tailored treatment planning. But the weak understanding of cell-of-origin and developmental mechanism for Group 3 and Group 4 has been a wall to new drug development.
2. Human Cerebellar Organoids as a New Experimental Platform
The team adopted an established protocol (developed by extending Muguruma et al., Cell Reports 2015) to differentiate healthy donor iPSCs into human cerebellar organoids. About 60-90 days of 3D culture produces the human-cerebellum-specific cell types:
- Lower rhombic lip-derived cells: candidate origin for WNT subtype.
- External granule layer GNPs: origin for SHH subtype.
- Posterior rhombic lip precursors: candidate origin for Group 4.
- Nestin+ neural stem cell population: candidate origin for Group 3.
- Purkinje cells, astrocytes, oligodendrocytes: surrounding microenvironment.
Into these organoids, CRISPR/Cas9 was used to introduce subtype-driver mutations (CTNNB1 activating mutations, PTCH1 loss-of-function, MYC amplification, OTX2 abnormalities, etc.) in a cell-type-specific manner, enabling real-time tracking of each subtype’s development.
3. WNT Subtype: Origin and Molecular Pathology Confirmed
CTNNB1 activating mutations introduced specifically into lower rhombic lip-derived cells produced organoids in which:
- An undifferentiated proliferative population expanded within 2-4 weeks.
- WNT-pathway downstream targets AXIN2, LGR5, ASCL2 were highly expressed.
- Morphology matched classical WNT medulloblastoma (small cells, prominent nucleoli, spindle-like organization).
- Markers: elevated DKK1 and GAB1, nuclear β-catenin — the clinical WNT-medulloblastoma diagnostic markers.
The same CTNNB1 mutation introduced into external granule layer GNPs did not form medulloblastoma-like tumors. The cell-of-origin hypothesis — that “the effect of a mutation depends on the cell type that receives it” — was directly confirmed in human-cell systems.
4. SHH Subtype: Validating the Vismodegib Response
PTCH1 loss-of-function in external granule layer GNPs faithfully reproduced classical SHH medulloblastoma. GLI1, GLI2, PTCH1 (feedback elevation), SMO-pathway activation, MYCN expression elevation, the typical “nodular desmoplastic” tissue morphology.
The team treated this organoid with Vismodegib (SMO inhibitor, Genentech/Roche, FDA-approved 2012). SHH-pathway signaling was completely suppressed; tumor cell proliferation stopped. This matches the clinically observed Vismodegib response in SHH medulloblastoma.
This validation matters. Vismodegib was first approved for basal cell carcinoma. Pediatric medulloblastoma indication expansion has progressed cautiously due to safety concerns in children (effects on growth plates). Establishing organoid-based drug-response validation creates the possibility of using it as a pre-treatment selection assay, broadening clinical translation.
5. Group 3: How MYC Reprograms Development
Group 3 medulloblastoma has the worst prognosis and is driven primarily by MYC amplification. The team succeeded in reproducing Group 3 medulloblastoma by overexpressing MYC in Nestin+ neural stem cells within organoids.
Principal findings:
- MYC does more than promote proliferation — it reprograms cell fate. When MYC overexpression occurs at a specific developmental stage, precursors that would normally differentiate into neurons are instead locked into “undifferentiated proliferative state.”
- Chromatin analysis showed MYC controls many super-enhancers, persistently opening proliferation-related loci.
- BET inhibitors (JQ1, OTX015) released this super-enhancer control, causing Group 3 organoid tumors to regress.
This finding raises the priority of BET inhibitors in Group 3 medulloblastoma drug development. BET inhibitors are being evaluated across solid tumors, with particular efficacy expected in MYC-driven cancers — and Group 3 medulloblastoma is the prototypical target.
6. Group 4: Unipolar Brush Cell Origin and the Role of OTX2
Group 4 is most frequent but with complex molecular pathology. OTX2 overexpression with KDM6A loss-of-function and other combinations occur. The team showed that OTX2 overexpression in unipolar brush cell precursors — a candidate Group 4 origin — produced Group 4-like tumors.
This adds a human-organoid evidentiary contribution to the long-running Group 4 cell-of-origin debate (cerebellar progenitor vs unipolar brush cell vs glutamatergic neuron precursor). Combining KDM6A loss-of-function produced more aggressive tumors with chromosomal abnormalities, reproducing within-Group-4 subset diversity.
7. Therapeutic Response: A “Personalized-Therapy Organoid” Proof of Concept
The team applied multiple treatments across the subtype organoids and compared response:
| Treatment | WNT | SHH | Group 3 | Group 4 |
|---|---|---|---|---|
| Standard chemotherapy (vincristine, cisplatin) | Strong | Moderate | Weak | Moderate |
| Vismodegib (SMO inhibitor) | None | Strong | None | None |
| BET inhibitors (JQ1, OTX015) | Moderate | Moderate | Strong | Moderate |
| mTOR inhibition (Rapamycin) | Moderate | Moderate | Strong | Moderate |
| WNT inhibition (PORCN inhibitor) | Strong | None | None | None |
This matrix matched clinically observed treatment responses by subtype. Organoid drug screening can serve as a pre-treatment “predictive assay”.
More importantly, this enables pre-screening of new drug candidates in human medulloblastoma-representative organoids. New BET inhibitors, CDK7 inhibitors, HDAC inhibitors, epigenetic-targeted drugs — all can now be screened in models that directly represent human medulloblastoma.
8. Limitations and Caveats
The paper’s strengths are large, but caveats remain.
First, organoid limitations. Organoids cannot reproduce the immune system, vasculature, or neural connectivity of human tumor microenvironment. CAR-T and other immunotherapy evaluations require alternative approaches.
Second, patient-derived organoid sample size. The paper’s main results use healthy-donor iPSCs + CRISPR mutations. Patient-derived organoids (PDOs) remain limited in number and cannot fully represent inter-patient diversity yet.
Third, long-term culture limits. 60-90 days of culture is substantial but may not fully reproduce the slow progression (months-years) of human medulloblastoma.
Fourth, cost and scalability. Bringing personalized-therapy organoid assays into clinical routine requires automation, standardization, and cost reduction.
Summary
- The April 2026 Cell paper successfully reproduced all four medulloblastoma subtypes (WNT, SHH, Group 3, Group 4) using human iPSC-derived cerebellar organoids + CRISPR mutation introduction.
- The cell of origin for each subtype (lower rhombic lip, external granule layer GNP, Nestin+ stem cells, unipolar brush cell precursors) was directly confirmed in human developmental biology.
- Subtype-specific drug response — Vismodegib (SMO inhibitor for SHH), BET inhibitors (for Group 3), PORCN inhibitor (for WNT) — matched clinical observations in organoid validation.
- Proof of concept for a personalized-therapy pre-treatment assay: “patient-specific medulloblastoma organoids predict treatment response.”
- Limitations: missing immune/vascular components, limited patient-derived organoid sample size, long-term culture limits, and clinical-routine cost-and-scale challenges.
My Thoughts and Outlook
The paper’s significance lies in providing — for medulloblastoma, a “treated but cognitively-scarring” pediatric cancer — a path toward “choosing more precise therapy and avoiding unnecessary toxicity.” Together with the progenitor-niche paper from Volume 1, this is one of the new-generation models of cancer origin reproduction in the laboratory.
Three structural implications for the global research and industry community. First, organoid platforms are becoming a foundational cancer drug-discovery infrastructure. The Wyss Institute (Harvard), the Tuveson Lab at Cold Spring Harbor (pancreatic cancer organoid pioneers), the Clevers Lab at Hubrecht Institute (Netherlands), the Salk Institute, and the Memorial Sloan Kettering Cell Engineering platform are all building independent organoid platforms. Pharmaceutical companies (Roche, Novartis, Pfizer) are internalizing organoid technology for drug pre-screening. Second, regulatory frameworks are evolving. The FDA has begun accepting organoid-derived efficacy data in IND submissions for select cases. The first medulloblastoma organoid-driven IND is likely within the next 5 years. Third, pediatric cancer demands special clinical platforms. Centers like St. Jude Children’s Research Hospital, Dana-Farber/Boston Children’s, and the Children’s Hospital of Philadelphia are positioned to integrate per-patient organoid assays with treatment selection. Multinational consortia for rare pediatric cancers (PNOC, PBTC) will likely adopt organoid pre-treatment assays as part of clinical trial protocols.
Volume 3 of this series turns to the Nature 2026 paper on epigenetic memory of colitis driving subsequent tumorigenesis — the third lens on cancer origin: not “mutation vs niche,” not “mouse vs organoid,” but “the memory of inflammation.”
Coming Next
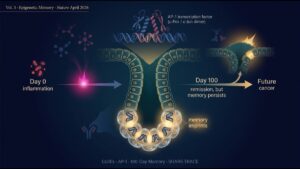
Volume 3 (final) examines the Nature 2026 paper by Nagaraja et al., “Epigenetic memory of colitis promotes tumour growth.” Chronic inflammation, even after clinical resolution, leaves a memory in colonic stem cell chromatin that persists for over 100 days, cooperating with subsequent oncogenic mutations to accelerate tumor formation. AP-1 transcription factors and SHARE-TRACE — a new single-cell lineage-tracing technology — illuminate the causal mechanism between chronic inflammation and cancer.
Edited by the Morningglorysciences team.


Comments