Key Takeaways
- The April 16, 2026 issue of Nature published a landmark paper by Nagaraja et al. (Harvard / Broad / MIT, Nature 2026;652:774) showing that chronic inflammation (colitis) leaves an “epigenetic memory” in the chromatin of colonic stem cells that persists for over 100 days after disease resolution. Following the progenitor niche (Vol. 1) and cerebellar organoid (Vol. 2) papers, this is the third lens on cancer origin: “the memory of inflammation.”
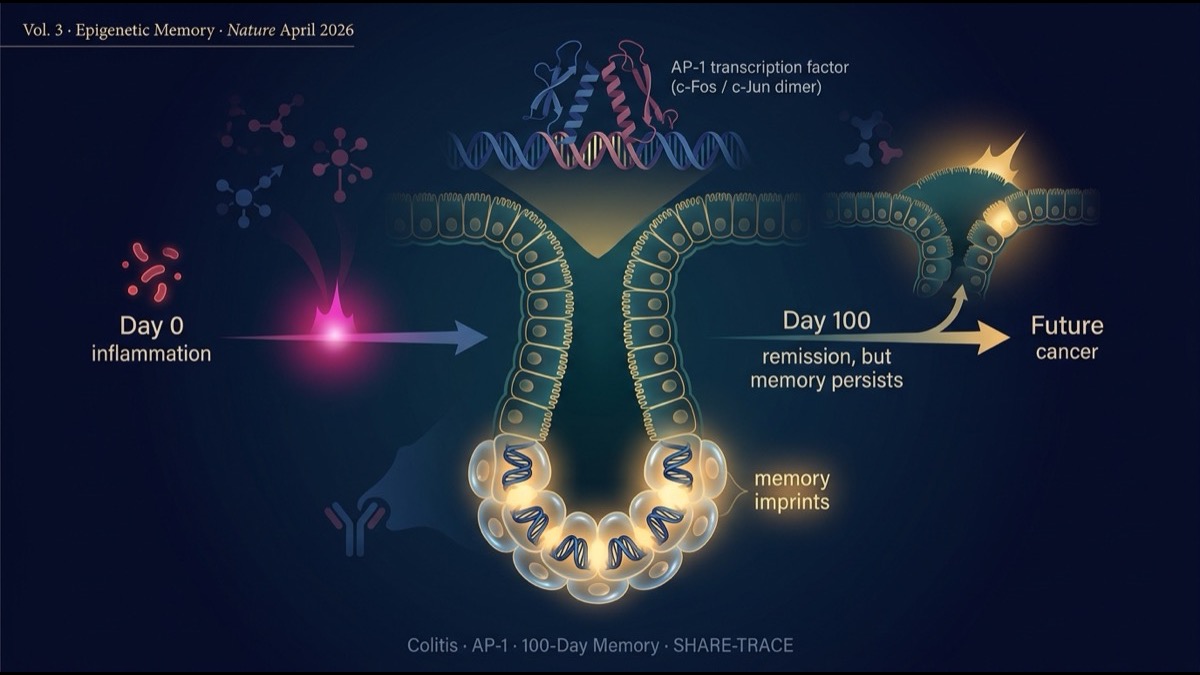
- Mechanistically, the core is the cumulative activity of the AP-1 transcription factor complex. During colitis, AP-1-dependent (c-Fos/c-Jun dimer) chromatin changes accumulate in colonic stem cells and persist for over 100 days after recovery. When subsequent oncogenic mutations (KRAS, APC, etc.) occur, this “memory” cooperates to accelerate tumor formation.
- The team developed SHARE-TRACE, a new single-cell technology that simultaneously profiles gene expression, chromatin accessibility, and clonal lineage in single cells. This enabled direct comparison of “clones with memory vs without memory,” and proved that epigenetic memory propagates clonally through stem cell divisions.
- Clinical implications: the elevated colorectal cancer risk in inflammatory bowel disease (IBD: ulcerative colitis, Crohn’s disease) patients has long been explained as a linear “inflammation → cancer” causality. This paper proposes a “memory of inflammation + subsequent mutation” cooperative model that may reshape IBD-associated cancer prevention. AP-1 inhibition emerges as a novel therapeutic axis.
Introduction — How Do You Measure “Inflammation Promotes Cancer”?
Chronic inflammation as a cancer risk factor is a long-known clinical observation:
- Chronic Helicobacter pylori infection → gastric cancer (5–10× risk)
- Chronic hepatitis B/C → hepatocellular carcinoma (10–100× risk)
- Inflammatory bowel disease (IBD: ulcerative colitis, Crohn’s disease) → colorectal cancer (2–5× risk, proportional to disease duration)
- Chronic pancreatitis → pancreatic cancer (elevated risk)
The traditional explanation: “inflammation directly damages DNA, mutations accumulate, cancer arises.” Reactive oxygen species (ROS)-induced DNA damage, direct mutagenicity of inflammatory cytokines, replication errors from increased cell division during regeneration — these are partially correct.
But the past five years of epigenetics research has surfaced an alternative mechanism: “inflammation alters not only the DNA sequence but how the genome is used.” Histone modifications, DNA methylation, chromatin accessibility shift — and even without mutations, gene expression changes in ways that drive cancer.
The April 2026 Nature paper by Nagaraja et al. reports the striking discovery that this “epigenetic effect of inflammation” is stored as long-term memory in stem cell chromatin. Using a new single-cell technology, SHARE-TRACE, the team causally demonstrated that this memory cooperates with subsequent oncogenic mutations.
This article walks through the paper’s strategy, principal results, mechanism, and clinical implications, with structural reflections for the global research and industry community.
Main Body
1. Epidemiology of IBD-Associated Colorectal Cancer
Quick recap of the clinical importance. Patients with ulcerative colitis or Crohn’s disease show colorectal cancer risk that increases with disease duration (Eaden et al. Gut 2001; Dyson et al. Aliment Pharmacol Ther 2012):
- 10 years of disease: ~2× risk
- 20 years: ~8× risk
- 30 years: ~18× risk
This is why IBD patients undergo regular colonoscopic surveillance. But the molecular mechanism for “why does risk scale with duration?” has been unclear. Classical explanation: cumulative inflammation → cumulative DNA damage → cumulative mutations. But this fails to fully explain a key clinical observation — risk persists even after inflammation has fully resolved. If inflammation has stopped, risk should stop scaling. Yet it persists. Something must be “remembered.”
2. Capturing “Memory” in a Mouse Colitis Model
The team used the dextran sodium sulfate (DSS)-induced colitis model. Mice receive DSS-water for 5 days, inducing acute colitis. Then over 100+ days on regular chow, inflammation fully resolves.
Measurements at three timepoints:
- Inflammatory phase: stem cells during acute colitis.
- Early remission: immediately post-resolution (10 days later).
- Long-term remission: after 100+ days, “no traces of inflammation.”
At each timepoint, colonic stem cells (Lgr5+) were isolated and profiled for chromatin accessibility (ATAC-seq), histone modifications (CUT&RUN), and transcriptome (RNA-seq).
3. Principal Finding 1: Chromatin Changes That Persist 100 Days
The result was striking. Stem cells 100 days after colitis appear externally indistinguishable from healthy mice. Histologically and clinically, inflammation is fully resolved. But genome-wide chromatin analysis revealed access patterns clearly different from inflammation-naive control stem cells:
- Inflammation-related loci maintained “open” chromatin states.
- Accessibility at NF-κB, AP-1, STAT-pathway target loci was elevated.
- For histone modifications, H3K27ac (active enhancer mark) was sustained at inflammation-related enhancers.
- At the gene-expression level, these genes were in a “primed for strong expression on demand” state.
Colonic stem cells had stored the inflammation experience “as memory in chromatin structure.” Quiet at baseline, but ready to respond strongly when stimulated.
4. AP-1 Is the “Enzyme That Writes Memory”
What molecule sits at the heart of this epigenetic memory? Through chromatin analyses and functional inhibition experiments, the team identified the AP-1 transcription factor complex (c-Fos / c-Jun dimer).
AP-1 is the classical “immediate early gene” transcription factor, rapidly activated by stress, inflammation, and growth factor stimulation. The findings:
- During colitis, AP-1 binding sites undergo widespread chromatin opening.
- After inflammation resolution, the “open” state at AP-1 binding sites is maintained.
- Nearby H3K27ac is sustained, leaving these sites functionally primed as enhancers.
- Genetic AP-1 inhibition (c-Fos/c-Jun loss-of-function) blocks memory formation.
AP-1 functions as the “enzyme that writes the inflammation experience into chromatin.”
5. SHARE-TRACE: A New Single-Cell Technology
The technical heart of the paper is proving that this memory “propagates clonally through stem cell divisions.” This required a new technology: SHARE-TRACE.
SHARE-TRACE simultaneously profiles, from a single cell:
- Transcriptome (analogous to scRNA-seq)
- Chromatin accessibility (analogous to scATAC-seq)
- Clonal lineage (CRISPR barcode tracing)
From this triple-channel data, one can directly assess “whether daughter cells from the same originating clone share inflammation-memory chromatin features.”
The result: daughter cells of the same originating clone share inflammation-memory chromatin features at high statistical frequency. Furthermore, memory strength varies among clones — “strongly remembering clones” coexist with “weakly remembering clones,” with strongly remembering clones inheriting their memory across generations.
6. Principal Finding 2: Memory Cooperates with Oncogenic Mutations
Memory alone does not produce cancer. The team measured cooperation between memory and mutation by introducing oncogenic mutations (APC loss-of-function) into memory-bearing mice:
- Memory (DSS-experienced) + APC mutation → markedly more and larger colonic tumors.
- No memory + APC mutation → normal-level tumor formation.
- Memory + AP-1 inhibition + APC mutation → tumor formation reduced to memory-naive level.
“Inflammation memory” alone does not cause cancer, but combined with subsequent oncogenic mutations, it markedly accelerates tumor formation. AP-1 inhibition eliminates this acceleration.
7. Mechanism: AP-1 Target Gene Programs Drive Acceleration
What is the molecular reason memory + mutation accelerates tumors? The team showed:
- In tumor cells from memory + APC-mutation mice, AP-1 target gene programs are markedly upregulated.
- Targets include proliferation (cyclin D1, Myc), epithelial-mesenchymal transition (Snail, Slug), and ECM remodeling (MMP9).
- APC mutation activates WNT/β-catenin to supply proliferation signaling. Simultaneously, memory chromatin keeps AP-1 target genes “open” for strong expression, accelerating proliferation.
This double-accelerator mechanism provides molecular explanation for the clinical observation that “IBD-associated cancers progress faster and more aggressively than typical sporadic colorectal cancer.”
8. Clinical Implications: AP-1 Inhibition as a New Therapeutic Axis
Three clinical implications:
First, IBD-associated cancer prevention strategy. For long-disease IBD patients, beyond regular surveillance, a new concept of “chemoprevention via AP-1 pathway suppression”. Direct AP-1 inhibitors are limited, but applied research is starting on existing drugs that indirectly suppress AP-1 (JAK inhibitors, melanocortin pathway activators).
Second, risk stratification of long-remission IBD patients. If biomarkers detecting “chromatin memory strength” are developed, “patients with strong memory” could be identified as high-risk, with more frequent surveillance or chemoprevention. Stool-derived DNA methylation assays and blood AP-1 target gene expression profiles are candidate technologies.
Third, a new therapeutic axis for established IBD-associated cancer. Even in patients with established cancer, AP-1 pathway inhibition may dismantle part of the proliferation drive. Therapeutic designs combining WNT pathway inhibition (PORCN inhibitors) become conceivable.
9. Limitations and Caveats
The paper has the following limitations:
First, mouse-to-human generalization. Whether mouse memory mechanisms function with the same intensity and persistence in humans requires chromatin analysis on serial biopsies from human IBD patients.
Second, AP-1 alone vs cooperation with multiple pathways. NF-κB, STAT3, HIF-1α and other inflammatory pathways may also contribute to epigenetic memory.
Third, generalization to other chronic inflammation–cancer relationships. Whether the same mechanism applies to Helicobacter pylori-gastric cancer, HBV/HCV-liver cancer, etc. AP-1 involvement is context-dependent.
Fourth, AP-1 inhibition toxicity concerns. AP-1 is essential for normal wound healing, immune response, and tissue regeneration. Chronic dosing toxicity needs careful evaluation.
Summary
- Nagaraja et al. Nature 2026 demonstrated that chronic colitis leaves an AP-1-dependent epigenetic memory in colonic stem cell chromatin that persists for over 100 days.
- The new technology SHARE-TRACE (triple single-cell profiling of transcriptome, chromatin, and lineage) proved that this memory propagates clonally through stem cell divisions.
- Memory alone does not cause cancer, but combined with subsequent oncogenic mutations (APC loss-of-function), it markedly accelerates tumor formation. AP-1 inhibition eliminates this acceleration.
- Mechanism: APC-mutation-driven WNT activation combined with memory-chromatin-driven strong expression of AP-1 target genes — a double-accelerator.
- Clinical applications: AP-1 inhibition chemoprevention, chromatin memory biomarkers, new therapeutic axis for established IBD-associated cancer.
- Limitations: mouse-to-human generalization, AP-1 alone vs multiple pathways, applicability to other cancers, AP-1 inhibition toxicity.
Series Synthesis
Across the three-volume series “Frontiers of Cancer Origin Research,” we have dissected three landmark papers from April 2026.
Volume 1: Reyes et al. (Cell) progenitor niche model — mutations alone don’t make cancer; specific progenitor populations form a “self-reinforcing niche” at the transition point. Structural insight.
Volume 2: Cell medulloblastoma cerebellar organoid paper — reproducing in human iPSC + CRISPR what mouse models cannot, a new-generation cancer-origin reproduction in the laboratory.
Volume 3 (this article): Nagaraja et al. (Nature) epigenetic memory paper — chronic inflammation leaves long-term memory in stem cell chromatin, cooperating with subsequent mutations. The third lens.
These three studies represent paradigm shifts on three axes — space (niche), time (development), and memory (inflammation traces). Common conclusion: “mutation accumulation alone is not sufficient for cancer.” Tissue-level dynamic states, specific developmental timing, traces of past experience — these non-mutational elements drive cancer initiation. This collection of discoveries structurally rewrites discussions of cancer prevention, early intervention, risk stratification, and new therapeutic targets.
My Thoughts and Outlook
As series synthesis, the largest implication is that “cancer-origin research is shifting from mutation-centric genetics to a ‘contextual oncology’ that integrates tissue, time, and experience.” Watching this field as a researcher and industry observer, I would say: the past 20 years were the era of DNA sequencing and mutation cataloging. The coming decade will be the era of integrating single-cell spatial omics, organoid models, and dynamic epigenetic analysis.
Three structural implications for the global research and industry community. First, single-cell spatial omics, organoid platforms, and epigenetic dynamic analysis are becoming foundational infrastructure for cancer drug discovery. Centers integrating all three (the Broad Institute, Sanger Institute, Karolinska, A*STAR Singapore, German Cancer Research Center DKFZ) are positioned to lead the next decade. Second, IBD-associated cancer prevention is an underbuilt category. The combination of AP-1 inhibition chemoprevention + chromatin memory biomarkers + risk stratification offers a multi-billion-dollar global opportunity that diagnostic-pharmaceutical partnerships (Genentech-Foundation Medicine model, Roche-mySugr-PathAI model) can pursue. Third, regulatory frameworks for chemoprevention are evolving. The FDA, EMA, and APAC regulators are increasingly accepting biomarker-stratified prevention trials. The first AP-1-inhibition chemoprevention trial in IBD is likely within the next 3-5 years.
2026 is the year AI is rapidly commoditizing knowledge work. Cancer origin research, requiring physical tissue specimens, advanced imaging hardware, and clinically integrated protocols, sits in the “AI cannot replace” zone. This series has spotlighted three discoveries that create new clinical workflows, new device categories, and new therapeutic indications that AI alone cannot produce. I hope to continue sharing this contextual-oncology revolution with the readers who completed the series.
Edited by the Morningglorysciences team.


Comments